
 Cushing sendromu: İnsidans; 1-2/100.000. Obez ve kontrolsüz diabetiklerde %3-4.
Cushing sendromu: İnsidans; 1-2/100.000. Obez ve kontrolsüz diabetiklerde %3-4.
Cushing sendromu tipleri
ACTH bağımlı: cushing hastalığı, ektopik ACTH sendromu, ektopik CRH sendromu. ACTH bağımsız: adrenal adenom, adrenal karsinom, primer pigmente noduler nodüler, adrenal hiperplazi ve carney sendromu.
Pseudo-cushing sendromu: alkolizm, depresyon, obezite.
Hiperkortizolemi etiolojisi
Pseudo-Cushing (nonpatolojik hiperkortizolemi): akut/kronik medikal hastalıklar, psikiyatrik hastalıklar, alkolizm.
Subklinik cushing sendromu: adrenal adenom (insidentoloma), adrenal makronodüler hiperplazi (nadir), hipofizer kortikotrop adenom (nadir),aberrant reseptör ekspresyonu (nadir).——-Cushing sendromu (patolojik hiperkortizolemi), dışarıdan GK kullanımı, topikal GK kullanımı (inhale, nazal,dermal), enjekte GK kullanımı (artiküler,periartiküler,intramüsküler),naturopatik preparasyon, endojen glukokortikoid üretimi.
ACTH bağımlı: hipofizer kortikotrop adenom, MEN1, hipofizer kortikotrop hiperplazi (CRH), ektopik ACTH sendromu (yulaf hücre Ca, bronşial, timik, splenik karsinoid,FEO, meduller tiroid Ca, adacık hücre tümörü).
ACTH bağımsız: adrenal adenom, adrenokortikal karsinom, mikronodüler ve makronodüler hiperplazi, aberrant reseptör ekspresyon, pigmente nodüler hiperplazi (carney sendromu), McCune Albright.
Subklinik cushing sendromu
İnsidental olarak adrenal kitle saptananların %5-20’inde HPA anormalliği ve otonomi gösterildi. Cushing sendrom bulgu ve laboratuvar belirtileri yok.Yüksek oranda hipertansiyon, İGT, DM, kardiyovasküler disfonksiyon var. Lezyon çıkarılırsa kaybolur. Aşikar cushing nadiren gelişir. Tedavi bireyselleştirilmelidir.
Cushing Sendromu Bulguları
Akne, hirşutizm, pletorik yüz, aydede yüz, pigmentasyon, KAH, KMP, hipertansiyon, skar pigmentasyonu, osteoporoz, morarma, ince deri, amenore, kas güçsüzlüğü, ülserasyonlar, ödem, supraklaviküler yağ yastığı, poliür,, böbrek taşı, stria, venöz tromboz.
Klinik özellikleri
Semptomlar: kilo artışı, adet düzensizliği, hirsutizm, psişik yakınmalar, sırt ağrısı, kas güçsüzlüğü.
Bulgular: obezite (trunkal, generalize), pletora, aydede yüzü, HT, deride bere, ekimoz, mor strialar, kas güçsüzlüğü, diabet (aşikar, IGT), osteoporoz, böbrek taşı, pigmentasyon.—–Klinik şunlara bağlıdır; glukokortikoid aşırılığı, mineralokortikoid artışı, androgenik hormon artışı, hipofiz tümörüne bağlı olarak gelişir.
Tanı ve ayırıcı tanı
Hiperkortizolemiye, buna yol açan nedenlere, lezyonun lokalizasyonuna bakılır.
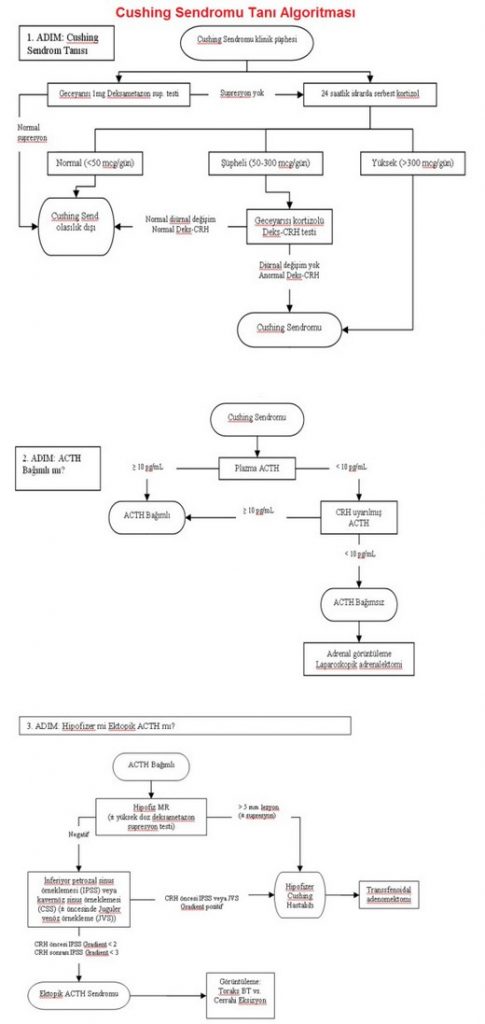
Cushing sendromunu tanımla: geceyarısı 1mg deksametazon supresyon testi,24 saatlik idrar serbest kortizol ölçümü, gece yarısı serum ya da tükrük kortizol ölçümü, deksametazon-süpresyon-CRH uyarı testi yapılır.
Hiperkortizolemiyi doğrulayan testler: kortizolün diürnal ritminin kaybı, idrar serbest kortizol atılımı ölçümü, gece yarısı dekzametazon süpresyon testi, iki günlük düşük doz dekzametazon testi, insülin hipoglisemisine kortizol yanıtı.———-Hiperkortizolemiye yol açan nedenlere şunlarla bakılır; ACTH ölçümü, yüksek doz dekzametazon süpresyon testi (bir gecelik – 8mg, iki günlük), metirapon testi, CRH testi, inferior petrozal sinus kan örneklemesi (İPSKÖ).
Geceyarısı 1 mg deksametazon süpresyon testi: sensitivitesi yüksek, spesifisitesi düşük, %5-30 yalancı pozitiflik vardır, 23.00’de 1mg verilir. Sabah 8.00’de serum kortizol <5 µg/dL ise tanı konur. Fenitoin, fenobarbital, rifambin, primidone (deksametazon metabolizmasını hızlandırarak), estrogen ve tamoksifen yalancı pozitifliliğe neden olur.
24 saatlik idrarda serbest kortizol ölçümü: 24 saatte >300 µg/dL tanıyı destekler. Yüksek volum ve tegretol (HPLC tekniği ile çapraz reaksiyon) kullanımında yalancı pozitiflik olur. Test 2-3 kez tekrarlanmalıdır.
Geceyarısı serum ve tükrük kortizol ölçümü: geceyarısı kortizol cushing sendromunda >7.5 µg/dL olur. Tükrük kortizolü evde yapılır. 0.2-0.55 µg/dL normal baskılanmayı gösterir.
Cushing Sendromu Tanı Algoritması
Lezyon lokalizasyonu
Adrenal USG, adrenal BT, hipofiz MRI, İPSKÖ, sintigrafi, tümör belirleyicileri lezyon lokalizasyonu amacıyla kullanılabilir.
Ektopik ACTH sendromu
Bronş karsinoid, küçük hücreli bronş kanseri, timik karsinoid, pankreas kanseri, pankreas adacık hücre tümörü, feokromositoma, over tümörü sebebiyle olabilir.
İdrar serbest kortizol: normali 20-90 µg/gün, cushingde >150 µg/gün.
1.0 dekzametazon süpresyon: Kortizol: normali <5 µg/dL, cushingde >10 µg/dL.
8.0 dekzametazonn süpresyon: Kortizol: Hipofizer ACTH bağımlı <%50 bazal, Ektopik -ACTH bağımsız >%50 bazal.
Cushing sendromu: Klinik şüphe > plazma bazal kortizol yüksek ve kortizol diurnal ritmi bozulmuş > gece yarısı tek doz dekzametazon testi sonu plazma sabah 8.00 kortizolüne bakılır > 5 µg/dL’den fazla ise > idrar serbest kortizole bakılır > düşük doz dekzametazon süpresyon testi (2 gün) ile kortizol ve insülin hipoglisemisine kortizol cevabı baskılanmaz ise > hiperkortizolemi denir.
Endogen cushing sendromu tedavisi: Otonom tümör dokusu rezeke edilmelidir. Cerrahi sonu stres doz GK kullanılır (hidrokortizon 20 mg X2-3/gün). Postop 2.gün sabah serum kortizol <2 µg/dL ise cerrahi cure öngörülür. GK kesildikten sonra sabah kortizol ve HPA incelenmelidir.
Cushing hastalığı tedavisi: Transfenoidal cerrahi sonu hiperkortizolemi varsa hipofizer irradiasyon endikedir.Hipopituitarizm, optik nöropati (nadir), beyin doku nekrozu ve sekonder beyin tümörü? gelişebilir. Ektopik tümör okkult ise bilateral adrenalektomi yapılır, GK ver MK replasmanı yapılmalıdır. Nelson sendromu gelişir (kortikotrop hücre tümörün agressif ve hızlı büyümesi).
Primer adrenal hastalık tedavisi: Laparoskobik yaklaşım esastır; adrenokortikal karsinom, koagulapati, daha önce cerrahi girişim ve travmada uygulanamaz. Noduler hiperplazi bilateral cerrahi ile tedavi edilir.
Cushing Sendromu Medikal Tedavisi
| Steroid biyosentez inhibitörleri | Etki mekanizması | Doz mg/gün | Etki % | Yan etki |
| Ketokonazol(Nizoral)Metyrapone(Metapirone)Aminoglutethimide (Cytadren) Mitotane (o,p’-DDD) (Lysodren) | Kortizol sentez inhibitörü11β-hidroksilaz bokeriKolesterolun pregnenelone dönüşümü blokeYan bağları bloke eder | 200-1200500-6000750-2000500-12000 | 7085>6083 | hepatotoksik, bulantı, ödem, jinekomasti, raş.Hirsutizm, akne, letarji, ataksi,ödem,bulantı,raşLetarji, somnolans, raş, ateş,bulantı, hipotiroidi, anorkesiya,GİS,hiperlipdemi, raş,ateş, jinekomasti,hepatotoksik |
| ACTH inhibitörleri | Etki mekanizması | Doz | Etki % | Yan etki |
| Siproheptadine(Periactin)Bromokriptine(Parlodel)Oktreotide (Sandostatin) Valproik asid (Depakene) | ACTH salınımı azalırACTH salınımı azalırACTH salınımı azalırGABA potansiyelize olur. ACTH ve CRH baskılanır | 24 mg/g3.75-30 mg/g100-600 µg/g1-2 g/g | 30-5025-42Sınırlı deneyimSınırlı deneyim | Hiperfaji, kilo, somnolansPostural hipotansiyon, kuru ağızDiare, safra taşıSedasyon, pankreatit, bulantı, hepatotoksik |
| Glukokortikoid reseptör antagonisti | Etki mekanizması | Doz kg/g | Etki | Yan etki |
| Mifepristone(RU-486, Mifeprex) | GlukokortikoidReseptör antagonisti | 10-25 | Sınırlı deneyim | Bulantı, kusma, düzensiz adet |
Primer adrenokortikal yetmezlik
Otoimmün adrenalitis >%80, infeksiyon >%20 (Tbc, histoplazmozis, blastomikozis, kriptokokkozis, bakteryel, HİV ve fırsatcı enfeksiyonlar), bilateral adrenal hemoraji (postop, travma, heparin), bilateral adrenalektomi, metastatik hastalıklar (akciğer, mide, meme), KAH, adrenolökodistrofi (X’e bağlı), kortizol biyosentezini azaltanlar (mitotane, ketokonazol, aminoglutethimid, metyrapon), kortizol metabolik klirensini hızlandıranlar (rifampin, fenitoin, fenobarbital).
Cushing hastalığı tedavisi
Transsfenoidal adenomektomi önerilir. Kavernoz sinüs invazyonu varsa transfrontal yaklaşım. İntraoperatif ultrason ya da MRİ tümör lokalizasyonu için gerek.Tümör lokalizasyonu yapılamadı ise hemihipofizektomi endikedir. Başarı %68.5-91, relaps 10 yılda >%15. Mortalite düşük. Menenjit, venöz tromboembolizm, hipopituitarizm, hemoraji, görme kaybı, likör sızması yan etkiler. Vazopressin salınım anomalisi postop olabilir, Dİ, SİADH gelişebilir. Postop 6.haftada hipofiz fonksiyonları incelenmelidir.
Hipoadrenalizm bulguları
Şuur kaybı, vertigo, konfüzyon, papil ödem, pigmentasyon, guatr, taşikardi, hipotansiyon, bulantı, kusma, diare, kas güçsüzlüğü, pigmentasyon, pigmente skar, pubik kıllanmanın azalması.
Sekonder adrenokortikal yetmezlik
1-İatrojenik; ekzogen glukokortikoid tedavinin kesilmesi, yada uzun süre CS alanlarda stres sırasında yeterli CS vermemek.
2-Tümörler; fonksiyone/nonfonksiyone hipofizer tümörler, kraniyofarengioma.
3-İzole ACTH yetersizliği.
4-Travma.
5-İnfarktüs/vasküler; iskemik nekroz, Scheehan sendromu, DM, orak hücre hastalığı, hipofizer apopleksi.
6-Hipofiz ışınlaması; fonksiyonlarda tedrici azalma.
7-İnfiltratif ve infeksiyöz hastalıklar; sarkoidozis, hemokromatozis, meningitis, tbc. 8-Otoimmün; lernfositik hipofizitis.
9-İdiyopatik.
Adrenal yetmezlik klinik özellikleri
Yakınmalar: iştahsızlık, yorgunluk, halsizlik, bulantı, kusma, konstipasyon, karın ağrısı, diare, tuz isteği, postural hipotansiyon, kas ve eklem ağrısı, aşırı tuz isteği, baş dönmesi. Belirtiler: kilo kaybı, hiperpigmentasyon, hipotansiyon, vitiligo.
Laboratuvar bulguları: hiponatremi, hiperpotasemi, hiperkalsemi, azotemi, anemi, eozinofili.——Kadınlarda adrenal androgen eksikliğine bağlı pubik ve aksiller kılların kaybı. Kitleye bağlı baş ağrısı, sinir paralizisi, dışarıdan CS kullananlarda cushingoid görüntü vardır.
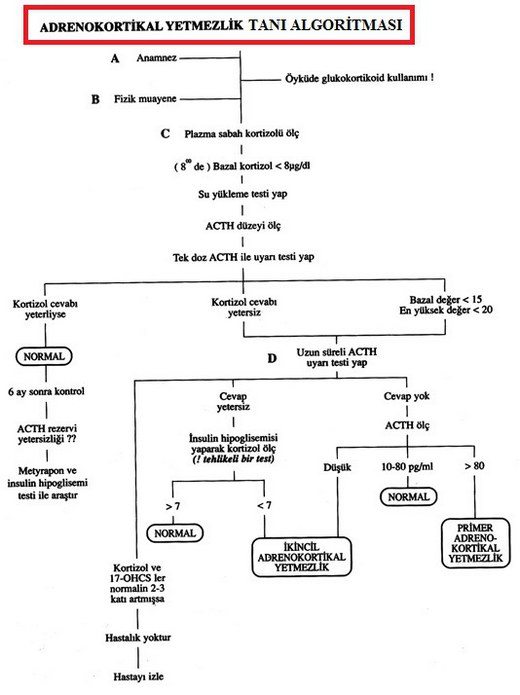
Primer adrenokortikal yetmezlik güncel tanı
Kortizol düşüktür. ACTH’a kortizol cevabı yetersizdir. Primerde ACTH düzeyleri artmış >200 pg/mL, sekonderde normal ya da azalmış 0-50 pg/mL. Primerde glukokortikoid ve mineralokortikoid düzeyleri azalmış,sekonderde sadece glukokortikoid yetersiz.
Adrenokortikal yetmezlik laboratuvar
Primerde ACTH yüksek + düşük kortizol vardır. Sekonderde ACTH normal ya da düşük + düşük kortizol vardır. Sabah kortizol ≤ 3µg/dL düşün, ≥ 19 µg/dL dışlanmalıdır. Dinamik test: cosyntropin; 250 µg İV-İM yapılır, 0,30,60 dakikalarda kortizola bakılır. Normal cevap 18 µg/dL. Beraberinde aldosteron 30. dakikada 16 µg/dL, sekonderde ACTH yetersizliğine bağlı adrenal atrofi vardır, kortizol <18 µg/dL. CT ve MRİ yararlıdır.
Adrenokortikal Yetmezlik Tanı Algoritması
Adrenokortikal yetmezlik tedavi
Akut: primer ve sekonder adrenokortikal yetmezlik tedavisinde; hidrokortizon Na suksinat verilir.
Kronik: Primer adrenokortikal yetmezlik tedavisinde; hidrokortizon, prednison, cortisone acetate, fludrokortizon verilebilir. Sekonderde fludrokortizonun yeri yoktur.
Hiperaldosteronizm
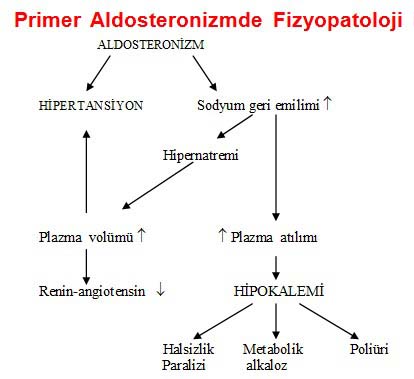
Hipermineralokortikoidizm (adrenal kökenli):
Aldosteron aşırılığı: aldosteran salgılayan adenom (%65), idiyopatik hiperaldosteronizm (%30-40), primer adrenal hiperplazi (<%1), glukokortikodle düzelebilen aldosteronizm %1-3, aldosteron salgılayan karsinom (%0,1-2).
Deoksikortikosteron aşırılığı: deoksikortikosteron salgılayan tümörler, KAH, 11ß- hidroksilaz eksikliği, 11a-hidroksilaz eksikliği.
Mineralokortikoid aktivite artışı (renal kökenli): Mineralokortikoid reseptörü aktive eden mutasyon, pseudohiperaldosteronizm tip II (Gordon), 11-hidroksisteroid dehidrogenaz eksikliği, doğumsal-görünürde minerakokortikod fazlalığı, edinsel (meyan kökü, karbenoksolon, Liddle sendromu).
Primer Aldosteronizmde Fizyopatoloji:
Primer aldosteronizm bulguları: Chvostek bulgusu, hipopotasemi EKG’si, çok nadiren adrenal tümör palpasyonu, kas güçsüzlüğü ya da paralizi, papil ödem (çok nadir), hipertansiyon, poliüri, glikoz, ödem (nadir).
Primer Aldosteronizm Tanı Algoritması:
Feokromositoma
Adrenal medullanın kromaffin hücre tümörüdür. Bu hücreler embriyogenik nöral krest kökenli, sempatetik orijinli. Abdomen ve toraks kökenli kromaffin hücre tümörlerine paraganglioma denir. Feokromositoma ve abdomen ve torakal paragangliomalar katekolamin (Cas) ve metabolitlerini sekrete ederler. Baş ve boyun kökenli paragangliomalar (en sık ckarotid cisimciği ve vagal, jugular ve timpanik ganglionlardan) parasempatetik kökenli ve Cas salgılamazlar. Hipertansif hastaların %0.1’inde olur.
Feokromositoma ve paragangliomada klinik bulgu ve belirtiler: baş ağrısı, terleme, çarpıntı, paroksismal hipertansiyon, anksiyete, bulantı, dispne, sersemlik, postural hipotansiyon, diare/kabızlık, ateş. FEO’da hipertansif retinopati, fasiyal hemangio, sütlü kahve lekesi, taşikardi, tremor görülebilir.
Von Hippel Lindau hastalığı ve FEO Birlikteliği
Tip 1 FEO: retinal ve SSS hemanjiyoblastom, renal kist ve karsinım, pankreas kist ve tümörü, epididimal kistadenom.
Tip 2 FEO: Tip 2A; retinal ve SSS hemanjiyoblastom, epididimal kistadenom. Tip 2b; retinal ve SSS hemanjiyoblastom, renal kist ve karsinım, pankreas kist ve tümörü, epididimal kistadenom. Tip 2C; sadece FEO.
FEO tarama testi uygulanacak hastalar: HT + epizodik baş ağrısı, çarpıntı,terleme, dirençli HT,kan basıncında oynamalar, aneztezi, cerrahi ya da angiografi sırasında açıklanamayan hipotansiyon, familyal sendromlar (MEN-2 nörofibriomatöz), adrenal insidentaloma, idiyopatik dilate KMP olanlarda FEO araştırılır.
Feokromositoma ve paragangliomada tanı: Tanı katekolamin ve metabolitlerinin kan ve idrarda ölçümü esasına dayanır. Baş ve boyun paragangliomalarda kitle palpe edilir. Sağırlık, disfaji ve disfoni vardır. En hassas yöntem plazma ve idrarda metanefrin düzeylerini ölçmektir. Klonidin süpresyon testi: 300 µg /gün klonidin artmış sempatik aktiviteyi baskılar. Feokromositomada plazma katekolamin düzeyleri azalmaz. Lokalizasyon; %90 adrenal kökenli. Ekstraadrenal en sık batında.
Tanıda şunlar önemlidir: plazma serbest metanefrin, idrar fraksiyone metanefrin, idrar katekolaminler, plazma katekolaminler, idrar total metanefrin, VMA.
Tümör lokalizasyonu: %90 adrenal glandda, ekstraadrenal tümör en sık abdomende, önce abdominal CT ya da NMRI önerilir. Tüm vücut sintigrafisinde 123I-MİBG kullanılır, spesifisitesi yüksek, sensitivitesi <%90. PET (pozitron emisyon tomografi) yararlı olabilir. Malignite tanısı metastaz varlığı ile konulur.
Feokromositomada tedavi
Küratif tedavi cerrahidir. İlk seçenek laparoskopidir. Bilateral feokromositoma cerrahisinde kronik hipokortizolemiden sakınılmalıdır. Cerrahi öncesi α-antagonistler verilmelidir; (Fenoksibenzamine = dibenzilin ve doksazosin = cardura) cerrahi sırasında katekolamin salınımına bağlı vazokonstruksiyonu bloke eder. Cerrahi sonu katekolamin düzeyleri ani azalır, reseptör down-regülasyonu vardır, volum kaybı gelişir, KB ciddi olarak düşebilir. Fenoksibenzamine; nonselektif, nonkompetitive α agonist, taşikardi yapar.Doksazosin; kompetetiive ve selektif α1bloker, taşikardi yapmaz.β-blokaj taşikardiyi azaltır. Ama α-blokaj sonu verilmelidir. KKB; nifedipine alternatif olarak kullanılabilir. Cerrahiden günler öncesi tuzlu su ya da plazma ile volüm ekspansiyonu sağlanır. Katekolamin salınımına neden olmayan inhale ya da İV aneztezikler kullanılır. Cerrahi sırasında ve sonunda kardiyovaskuler parametreler incelenir. Hipertansif nöbet; İV phentholamine (regitine), magnezyum sulfat, nitropurisside (nitropres) ile tedavi edilir. Aritmiler İV lidokain ya da kısa etkili β-blokerle tedavi edilir. Malign FEO’da α-metil-p-L-tyrosine (demser; katokolamin sentezini azaltır), α-antagonist, kemoterapi, 131I-MİBG kullanılabilir. Cerrahiden sonra ilk 24-48 saat içerisinde hipoglisemi ve hipotansiyona dikkat edilmelidir. Gebelikjte feokromositoma preeklampsi ile karışır. Trimestırde paroksizim yapar. fenoksibenzamin kullanılabilir. Laparoskopik olarak 2. trimestırde çıkartılmalıdır.
Cinsel farklılaşma bozuklukları
Bireyin dişi ve erkek oluşunu belirleyen faktörler: kromozomal seks,gonadal seks, fenotip, hormonal durum, psikojenik davranış.
Kromozomal seks: Ovumda 22+X kromozomu, spermde 22+X/Y kromozomu, zigot 46 XX, ya da 46 XY’dir. En sık rastlanan seks kromozom anomalileri; 45 X (turner sendromu), 47 XXY (klinifelter sendromu), 47 XXX, 47 XYY, seks kromozomlarında delasyon, 45 X/46Y mozaik formlar.
Gonadal seks: Gonadal seksi belirleyen seks kromozomlarıdır. Primitif gonad seks kromozomları etkisi ile over ya da testise dönüşür. Y kromozomu belirleyicidir. Y kromozomu yoksa gonad overe doğru gelişir.
Embriyojenik yaşamda cinsel gelişme: İn utero 9. haftadan sonra gonadlar testis ve overe dönüşür. İç genitalyayı belirleyen testislerdir. Müller kanalı dişi iç genitalyayı yapar; tubalar, uterus ve vajinanın 1/3 bölümü. MİF (sertoli hücre kaynaklı?) ile Müller kanalı geriler. Wolff kanalı gelişir. Wolff kanalından vas deferens, epididim, vezikula seminalis ortaya çıkar.
Eksternal dış genitalya: Hormonal etki ile olur. Gonadların salgıladığı testosteronla dış genitalya erkek tipini alır.
Konjenital adrenal hiperplazi: Kortizol biyosentezinde gerekli olan enzimlerden birinin kısmen/tümüyle yok olmasıdır. Tipleri; 21-hidroksilaz, 11β-hidroksilaz, 17α-hidroksilaz, 3β-ol-dehidrogenaz, D4-5-izomeraz.
21-hidroksilaz eksikliği: En sık görülen KAH formudur. Ağır form; doğumdan hemen sonra MK eksikliğine bağlı tuz kaybı + garip dış genitalya vardır. Hafif form; kızda virilizasyon, erkekte pseudo-prekoks puberte vardır. Doğumda kız çocuğu virilizasyon nedeniyle erkek kabul edilebilir. Dış genitalya erkek yapıda ama testisler yok. Boy hızla uzar sonra kısa kalır. En sık formu erişkin yaşa gelip de amenoresi olan kadındır. Klitoris büyük, sekonder seks karakterleri gelişmemiş, virilizasyon var. Ses kalın olabilir. Tanı: kanda 17-0H progesteron, idrarda metaboliti pregnanetriol artmıştır, ACTH artmıştır, androgenler ve idrarda 17-ketosteroidler artmıştır.
KAH’da tedavi amaçları: Epifiz kıkırdaklarının kapanmasını sağlamak. Aşırı androgenlerin gonadotropinler üzerine yaptığı baskıyı ortadan kaldırırarak normal puberteyi sağlamak. Kadında virilizasyonu önlemek, göğüs gelişmesini ve menstruasyonu başlatmak. Sürrenal yetmezliği ortadan kaldırmak. Prednizolone replasman için uygundur.
Cinsel farklılaşma bozuklukları:
1. Gonadların farklılaşmasında ortaya çıkan bozukluklar: turner sendromu, klinefelter sendromu, gonadal disgenezi, gerçek hermafroditizm.
2. Kadın yalancı hermafroditizmi (46 XX): KAH, gebelikte annenin androjen veya sentetik progesteron kullanması.
Geç başlayan 21-0H eksikliği: Konjenitaldir. Hafif enzim defekti, yakınmalar puberteden sonra olur. Menstruasyonun bozulması ve hirsutizmin ortaya çıkışı ile kendini gösterir. 17-OH progesteron normal, ACTH’ya yanıt abartılıdır.
11β-hidroksilaz eksikliği: 11β-hidroksilaz eksikliği nedeni ile kortizol yapımı azalır. ACTH artar. Zayıf mineralokortikoid aktivitesi olan 11-deoksikortikosteron yapımı artar. Androgen fazlalığı + hipertansiyon, hipopotasemi vardır. İdrarda 11-deoksikortizol ve metaboliti tetrahidro-11-deoksikortizol atılımı artar.
17α-hidroksilaz eksikliği: Pregnenolonun 17OH-pregnelone; progesteronun 17OH-progesterona dönüşümü defektif. Kortizol ve androjen yapımı azalmış. ACTH artmıştır, mineralokortikoid yolak işler. HT + hipopotasemi vardır. Kadında amenore, erkekte kadın fenotipi var. 17-ketosteroid ve 17-hidroksisteroid atılımı azalmıştır.
3β-ol dehidrogenaz eksikliği: Glukokortikoid, mineralokortikoid ve seks steroidleri azalır.Yeni doğanda tuz kaybı ve sürrenal yetmezliği var. DHEA aşırı yapılır (zayıf androgenik etki). Kız çocuğunda virilizasyon vardır. Erkek çocukta kadın fenotipi var. İdrarda 17-ketosteroidler ve DHEA miktarı çok artmıştır.
Erkek yalancı hermafroditizmi (46 XY):
1-Testosteron sentezi için gerekli enzim eksikliği.
2-Testosteron etkisizliği; testiküler feminizasyon, reinfenstein sendromu, 5α-redüktaz eksikliği.
3-Leydig hücre agenezisi; HCG ve LH’a testisler yanıtsız.
4-Erkek turner sendromu; 45 X, 46 XY.
5-Kaybolan testis; Vanishing testis.
6-MİF’in etkisizliği sonu iç genitalyanın kadın tipi olması.
7-In utero hayatta annenin estrojen ve progestojen alması.
Turner sendromu: Karyogram 45 XO. İç ve gış genitalya kadın tipinde. Boy kısa, boyun kısa ve kalın (webbed neck). Bilateral kubitus valgus vardır. Göğüs geniş ve meme başları uzak (shield chest). Meme dokusu gelişmemiş. Primer amenore vardır. Ellerde 4. metakarp kısa. KVH anomalileri sık iskelet anomalileri, renk körlüğü olabilir. Ekstremitede lenfödem olabilir. Zeka normal veya normalin alt sınırında olabilir. Tedavide siklik estrojen ve progesteron verilmelidir.
Klinefelter sendromu: Karyogram 47 XXY. Erkek fenotipi özelliği gösterir. Önikoid görüntü vardır.Testisler küçük ve sert, jinekomasti vardır. İskelet anomalileri olabilir. FSH ve LH artar, testosteron azalır. Tedavi testosteron/ömür boyu.
Gerçek hermafroditizm: Karyogram 46 XX,46 XX/46 Xy veya 46 XY olabilir. Tek tarafta testis ya da over, diğer tarafta ovatestis olabilir. Kriptorşidi mevcut. Jinekomasti olabilir, yarısında menstruasyon olabilir, ovulasyon, spermatogenez olabilir.
Testiküler feminizasyon: Androgene direnç var. Androgenle reseptör arasında ilişki bozuktur. Fenotipleri kadın. Karyotipleri XY. X’e bağlı geçiş gösterir. Pubertede seks karakterleri gelişir. Menarş olmaz.Kıllanma yoktur. Dış genitalya kadın tipindedir. İç genitalya erkek tipindedir.Testisler karın içi, kasık yada labyalarda. Testosteron artmıştır, estrojen artmıştır, LH artmıştır, FSH normal yada artmıştır. Testisler çıkartılır, estrojen tedavisine başlanır.
Reifenstein sendromu: Parsiyel androjen rezistansı (reseptöre bağlanma dokuduan dokuya farklı). Karyotip 46 XY. X’e bağlı resesif geçer. Gonadlar testisdir, dış genitalya erkek. İç genitalya wollf kanalında gelişir, hipoplazikdir.Testisler ve fallus küçük kalır.Azospermi vardır. Puberte sırasında jinekomasti ortaya çıkar. Testosteron artmıştır, LH artmıştır, FSH normal ya da artmıştır.Tedavi erken dönemde tanı konulur ise kadın olarak yetiştir.
5α-redüktaz eksikliği: Karyotip 46 XY. Otosomal resesif geçer. Testosteron dihidrotestosterona dönüşemez. İç genital organlar Wollf kanalından köken alır. Dış genitalya kadın tipinde. Kız çocuğu olarak yetiştirilirken pubertede kas yapısı güçlenir, ses kalınlaşır, klitoris büyür. Jinekomasti olmaz. Hasta kendini erkek hisseder. Spermatogenez durmuştur. Leydig hücreleri hiperplazikdir. Kıllanma çok azdır. Dihidrotestosteron düzeyleri kanda çok azalmıştır, LH artmıştır. Tedavide; hasta, kadın olarak yetiştirilir.
YAZIYI PAYLAŞ
Yazar Hakkında
Dr. Enes Başak
Aile Hekimliği
 Adrenal bez hastalıkları tanı ve tedavisi
Adrenal bez hastalıkları tanı ve tedavisi Cushing sendromu nedir? Nedenleri, belirtileri ve tedavisi
Cushing sendromu nedir? Nedenleri, belirtileri ve tedavisi Kortizol (stres hormonu) nedir? Ne işe yarar? Yüksekliği ve düşüklüğü
Kortizol (stres hormonu) nedir? Ne işe yarar? Yüksekliği ve düşüklüğü Cinselliği olumsuz etkileyen faktörler ve evliliği renklendiren 10 seks önerisi
Cinselliği olumsuz etkileyen faktörler ve evliliği renklendiren 10 seks önerisi Endokrin acillerin tanı ve tedavi yöntemleri
Endokrin acillerin tanı ve tedavi yöntemleri
YORUMUNUZ VAR MI?