
Nadir Hastalıklar: Tanıya giden uzun yol ve beklenen yeni tedavi seçenekleri

Dünyada etkilenen sayısı birkaç yüz kişiden üç dört kişiye kadar değişen çok nadir hastalıklar vardır. Bu hastalıklar her ne kadar çok nadir görülse de, bu tür hastalıkların çeşidi o kadar çok fazladır ki, çok nadir görülmelerine rağmen dünya çapında milyonlarca insanın etkilenmesine sebep olmaktadır. İnanılmaz derecede nadir görülen bu hastalıklardan muzdarip olanların hastalığı ya yıllarca hiç fark edilmez ya da yanlış teşhis konulduğu için doğru tedavi şansları olmaz. Bu nedenle çoğu hasta semptomlarının belirsizliği ile yaşamak zorunda kalır.
Bu hastalıkların nadir görülmesi doğal olarak araştırmalara çok az para ayrılmasına ve doğal olarak da ilaç ve tedavi olanaklarının çok kısıtlı olmasına sebep olmaktadır. Bu duruma dikkat çekmek için yılın en nadir günü olan 29 Şubat Nadir Hastalıklar Günü ilan edilmiştir.
Tanıya giden uzun yol
Bir hastalığın nadir hastalıklar kategorisine girme kriterleri ülkeden ülkeye değişmektedir. Örneğin Avrupa Birliği’nde bir hastalık 10.000 kişide 5’den daha az ise nadir hastalık olarak kabul edilir. Bu ilk bakışta az gibi görülse de, dünyada 6.000-8.000 arası farklı nadir hastalık olduğu düşünülürse toplam hasta sayısının hiçte az olmadığı görülecektir.
Türkiyede nadir hastalıklardan muzdarip hasta sayısının 5 milyon, dünyada ise 350 milyon kişi olduğu tahmin ediliyor. Toplam hasta sayısı bu kadar fazla olmasına rağmen, nadir hastalıklar konusu gerek toplumda gerekse tıpta genellikle pek önemsenmez. Bu nedenle, bu hastalıklara tıpta orphan diseases (yetim hastalıklar) etkilenenlere ise orphans of medicine (tıp yetimleri) denir [1 2 3 4 ].
Nadir hastalıkların teşhisi
Nadir hastalıkların belirtileri genellikle çocuklukta ortaya çıkar ve doktordan doktora yapılan başarısız muayeneler genellikle hasta için ek bir psikolojik yüktür. Bu tür hastalıklara doğru teşhis konması ortalama beş yıl sürmekte ve hastalar bu süre içerisinde ortalama sekiz kadar farklı doktoru ziyaret etmektedir.
Nadir hastalıkların semptomlarının çeşitli ve karmaşık olması ve hastalıklarla ilgili kapsamlı bir veritabanı olmaması, doktorların genellikle bu hastalıklara doğru teşhis koymasını zorlaştırmaktadır. Semptomların diğer klinik bulgularla benzerliği hastaların en az yarısına yanlış tanı konulmasına sebep olmaktadır. Hastalar kimi zaman ciddiye alınmazken, kimi zaman olağandışı semptomları uydurmakla suçlanırlar.
Nadir hastalıkların farklı nedenleri vardır
Nadir hastalıkların yüzde 80’nin sebebi genetik olduğu için belirtileri genellikle doğumdan kısa bir süre sonra ortaya çıkar ve ömür boyu sürerler. Bazı durumlarda ise virüs enfeksiyonları tarafından da tetiklenir.
I- Genetik mutasyonlardan kaynaklananlar
Nadir hastalıkların nedenleri çoğu zaman ilgili gende meydana gelen bir mutasyondan kaynaklanmaktadır. Bu mutasyonlar kimi zaman bir nükleotidin değişmesi veya silinmesi veya iki nükleotid arasına fazladan bir nükleotid eklenmesi şeklindedir. Bu durumda genin ifadesi değişerek kodladığı protein ya üretilemez ya da kusurlu üretilir. Bu da vücutta yıkıcı etkilere sebep olur.
II- Tipik bulaşıcı ajanlardan kaynaklananlar
- Viral enfeksiyonlar: Genetik nedenler kadar sık olmasa da patojenlerin sebep olduğu enfeksiyonlardan kaynaklanan nadir hastalıklar da vardır. Bunun iyi bilinen örneği kuduzdur. Viral zoonoz ile enfekte bir köpeğin ısırması ile insana bulaşan bu hastalık genellikle ölümcül olan ensefalitiyi (beyin iltihabı) tetikler. Bu tür ısırıklardan sonra profilaktik olarak pasif bir aşı uygulanarak kuduzun ölümcül etkisinden kurtulunur.
- Bakteriyel enfeksiyonlar: Bakteriyel enfeksiyonlardan kaynaklanan nadir hastalıklar da vardır. Örneğin Clostridium botulinum bakterisinin sebep olduğu Botulizm Bozuk gıdalar yolu ile alınan bu bakteri ölümcül gıda zehirlenmesine sebep olur. Bu bakteri, insanlarda nörotransmitter asetilkolini inhibe eder ve böylece sinirler ve kaslar arasındaki sinyal iletimini engeller. Güçlü zehirlenmelerde hasta tedavi edilmediği takdirde yüzde 90 oranında ölümle sonuçlanır. Hafif zehirlenme vakalarında ilgisiz birçok semptom nedeniyle tanı genellikle zordur. Bazen yanlış tanı da konabilir.
III- Çevresel faktörlerden kaynaklananlar
- Asbest: Nadir görülen hastalıkları etkileyebilecek başka bir sebep de endüstriyel maddelerdir. Bunların en meşhurlarından biri asbest Mikroskobik asbest lifleri çok dirençli mineraldir ve mezotelyoma olarak adlandırılan akciğer kanserine ve plevra kanserine neden olabilir. Milyonda sadece 1 ila 30 kişiyi etkileyen bu kanser türünün tedavisi zordur.
- Cıva: Cıva zehirlenmesi de nadir görülen hastalıklardan biridir. Ağız ya da burun yolu ile akciğerler veya mideye gelen cıva ciddi zehirlenmelere sebep olur. Cıva, beyni kan dolaşımındaki patojenlerden ve toksinlerden koruyan kan-beyin bariyerinden geçerek beyne de girebilir. Böyle durumlarda, beyindeki proteinlerin yapısı ve işlevi için gerekli olan kovalent bağların yapısı bozulur. Bu da özellikle sinir hücrelerinin işlevlerini yerine getirmesine engel olur.
En nadir görülen hastalıklar
Nadir hastalıklar arasında son derece olağandışı semptomlarla şaşırtan bazı hastalıklar da vardır. Çok nadir olmaları nedeniyle öne çıkan bu hastalıklardan bazıları dünya çapında yalnızca birkaç yüz kişiyi etkilerken, bazıları sadece onlarlarca kişiyi etkiler. Aşağıda en nadir görülenlerden beş örnek yer almaktadır.
Fibrodisplazi ossificans progresif (FOB)
Dünya çapında yaygınlığı yaklaşık 1: 2.000.000 olan bu hastalık tüm etnik grupları, coğrafi bölgeleri ve her iki cinsiyeti eşit olarak etkilenir. Bu hastalık bağ ve kas dokusunun yapısı kemiğe dönüşererek hareketi imkansız hale getirir.

Tabiri caizse, iskeletin üzerinde ikinci bir iskelet daha oluşur. Yeni doku hücreleri yerine yeni kemik dokusu oluştuğundan, en küçük yaralanmalar bile ciddi sağlık sorunlarının ortaya çıkmasına sebep olur. Şimdiye kadar, dünya çapında yaklaşık 800 akut vaka bilinmektedir.
Teşhis: Heterotopik kemikleşme (birbiri içinde) basit bir röntgen filmi çekimi ile teşhis edilebilir. Teşhisi doğrulamak için genetik testler yapılabilir. Çoğu FOP vakası sporadiktir (kalıtsal olmayan mutasyonlar). Az sayıda vaka kalıtsal mutasyonlar taşır. Kalıtım otozomal dominant tır.
Sebep: Hastalığa, ACVR1/ALK2 genin 2q24.1 lokasyonunda meydana gelen bir mutasyon sebep olur. Bu mutasyon ACVR1/ALK2 geni nin 617. pozisyonundaki Guanin’in Adenin ile değişmesi şeklindedir (617G>A; R206H). Bu mutasyon, ilgili proteinin hatalı kodlanmasına sebep olur ki, bu da artan kemik büyümesine yol açar [5].

Tedavi: FOP için şimdilik rutin sayılabilecek güvenilir bir tedavi yok ancak ilk belirtilerin ortaya çıktığı ilk 24 saati içinde dört günlük yüksek dozda kortikosteroid tedavisi uygulanırsa, hastalığın erken evrelerinde görülen şiddetli inflamasyon ve doku ödemi azalır [6] [7].
Yaşam beklentisi: Bir çok FOP hastasında ortalama yaşam süresi 40 yıldır. Çoğu hasta yirmili yaşların sonuna doğru tekerlekli sandalyeye mahkum olur. En yaygın ölüm nedeni, toraksın (kaburgalar ile çevrilen ve göğüs boşluğu) kısıtlı hareketliliğinden kaynaklanan komplikasyonlardır.
2- Erken Yaşlanma Hastalığı (Hutchinson-Gilford progeria)
Hutchinson-Gilford progeria sendromu, çocuklukta başlayan ölümcül, otozomal dominant erken yaşlanma hastalığıdır. Hastalık, büyüme geriliği, gelişme geriliği, tipik yüz görünümü (çıkıntılı alın, çıkık gözler, ince kemerli burun) ile karakterizedir. Hastalar ayrıca yetersiz alt çene gelişimi ve çıkıntılı kulaklar ve ciltte belirgin dermatolojik özellikler ile belirgin kutanöz damar sistemi gibi dışarıdan kolayca fark edilebilecek fiziksel özelliklere sahiptir [8].

Etkilenen çocuklar doğumda ve erken bebeklik döneminde normal görünürler ancak daha sonra akranlarından daha yavaş büyürler ve beklenen oranda kilo alamazlar. Bu durum entelektüel gelişimi, oturma, ayakta durma ve yürüme gibi motor becerilerin gelişimini etkilemez. Çocukluktan itibaren başlayan arterlerde ciddi sertleşme (ateroskleroz) görülür. Bu durum, genç yaşta kalp krizi veya felç geçirme olasılığını büyük oranda artırır. Bu komplikasyonlar zamanla kötüleşebilir ve yaşamı tehdit edebilir.

Hutchinson-Gilford progeria sendromu, dünya çapında 4 milyon yenidoğan çocukta 1 görülen çok nadir bir hastalıktır. Hastalık ilk kez 1886’da tanımlanmasından bu yana bilimsel literatüre 130 civarında vaka bildirilmiştir.
Başlıca belirgin özellikler
- Ortalamanın altında boy, kilo ve yavaş büyüme
- Dar yüz, küçük alt çene, ince dudaklar ve gaga şeklinde burun
- Yüz göre orantısız büyük bir kafa
- Belirgin gözler ve göz kapaklarının tam kapanmaması
- Kirpikler ve kaşlar dahil saç dökülmesi
- İncelme, sivilceli, buruşuk cilt
- Görünür damarlar
- Tiz ses
- Şiddetli ilerleyen kalp ve damarı hastalıkları
- Gövde ve ekstremitelerde cildin sertleşmesi ve sıkılaşması
- Gecikmiş ve anormal diş oluşumu
- Bazen işitme kaybı
- Deri altında yağ kaybı ve kas kütlesi kaybı
- İskelet anormallikleri ve kırılgan kemikler
- Sert eklemler
- Kalça çıkığı
- İnsülin direnci
Sebep: Hastalığa LMNA geninde meydana gelen mutasyonlar neden olur. LMNA geni, lamin A adında bir protein kodlar ve bu protein hücre zarının bileşenidir. Mutasyon nedeni ile hatalı üretilen lamin A proteini hücre zarını kararsız hale getirerek hücre çekirdeğinin zarar görmesine sebep olur. Sonuç olarak, DNA replikasyonu gibi sayısız hücre bölünme süreci artık doğru şekilde çalışamaz.
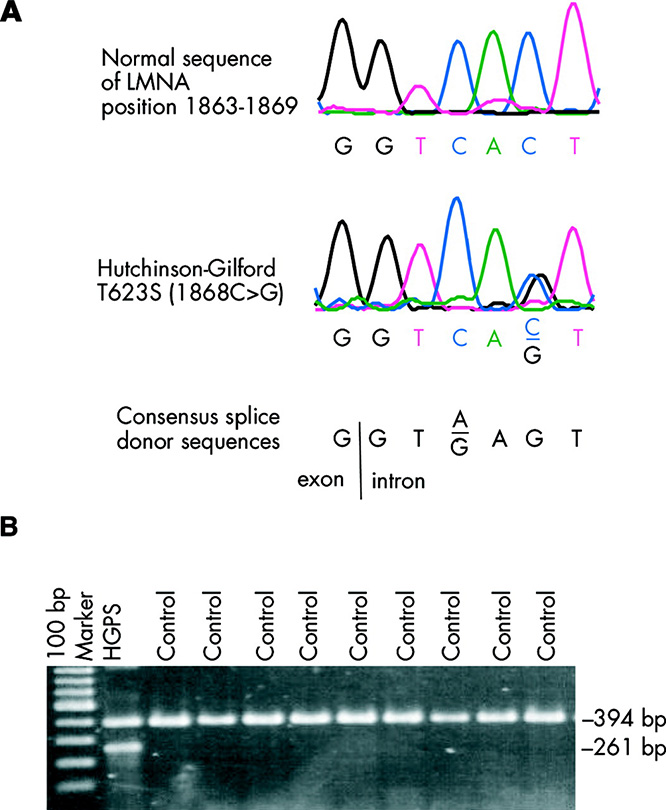
Bu da erken yaşlanmanın yolunu açar. Progeria, birçok genetik mutasyonun aksine nadiren ailelerde geçer. Vakaların bir çoğunda mutasyonu tesadüfen olan bir olaydır. Progerialı bir çocuğu olan ebeveynler için, ikinci bir progerialı çocuğa sahip olma olasılığı yaklaşık yüzde 2 ila 3’tür [9].
Tedavi: Kalıcı bir tedavi şimdilik mümkün değil ancak devam eden araştırmalar tedavi için umut vaat etmektedir. Profilaksi tedaviler ile komplikasyonlar azaltılabilir. Farnesil transferaz inhibitörü (lonafarnib) ile hastalığın ilerlemesi bir miktar geciktirilebilir.
Yaşam beklentisi: Progerialı çocukların çoğunun ölüm nedeni kalp problemleri veya felçtir ve ortalama yaşam beklentisi yaklaşık 13 yıldır. Bazı hastalar daha genç ölebilir, bazıları ise 20 yıl kadar yaşayabilir.
3- Desmoplastik yuvarlak hücreli tümör (Desmoplastic small round cell tumors DSRCT)
Desmoplastik yuvarlak hücreli tümör (Desmoplastic small round cell tumors DSRCT) 1989’da ilk kez tanımlandığından bu yana dünya çapında yalnızca birkaç yüz vaka rapor edilmiştir. Son derece nadir görülen bir kanser türdür. Tümör öncelikle karın içini kaplayan karın zarında (Periton) oluşur ve daha sonra lenf düğümlerine ve nihai olarak karaciğere yayılır. Tümör, ilk keşfedildiğinde çoğu zaman geç kalınmış ve çok büyümüş olur. Tümör radyasyon ve kemoterapi tedavisine dirençlidir.
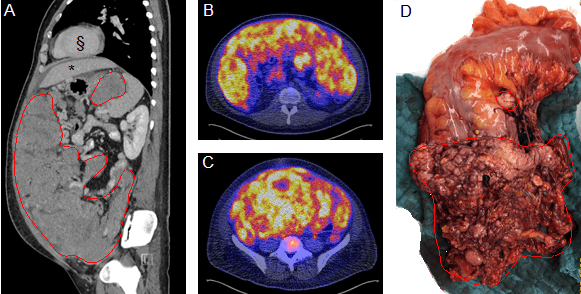
DSRCT, birçok insanda, başlangıçta semptom göstermez. İlk belirtiler tümör iyice büyüdükten sonra başlar. Teşhis, ultrason, BT, MRI ve PET gibi görüntüleme ve tarama teknikleri kullanılarak yapılır ve ayrıca biyopsi ile kanserin varlığı teyid edilir. [10]
Başlıca belirtiler
- Ağrı
- Mide bulantısı ve kusma
- İshal
- Kabızlık
- Karında şişlik
Sebep: Bu nadir hastalığın nedeni genetiktir. Hastalık hemen hemen tüm vakalarda, 11. ve 22. Koromozomlar arasında bazı lokasyonların değişiminden yani translokasyonundan (t(11;22)(p13;q12) kaynaklanır. Değişen kısımlar içinde EWSR1 ve WT1 genlerinin parçaları yer almaktadır. Bu genlerin tümör gelişimi ve kontrolsüz büyümeyi kontrol ettiği varsayılmaktadır. Belirlenen bu translokasyon dışında DSRCT ile ilişkili olan başka translokasyonlar da rapor edilmiştir. Bunlar şunlardır: (t(5;19), t(X;16) ve t(4;10))
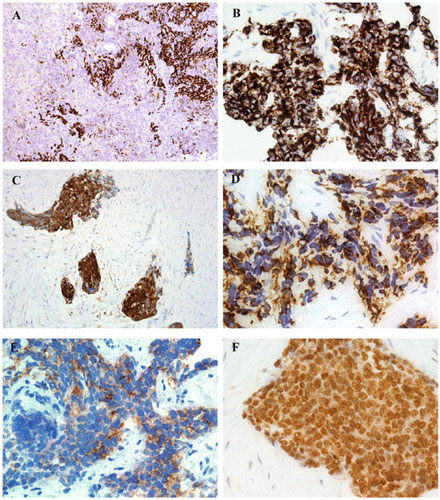
Tedavi: DSRCT, günümüzde agresif, multimodal bir terapi konsepti ile tedavi edilmektedir. Multimodal, farklı terapötik yaklaşımların birbiriyle birleştirilmesi ve birkaç tıp disiplininin el ele çalışması anlamına gelir. DSRCT’li hastalar genellikle önce kemoterapi alırlar. Mümkünse tümör dokusunun tamamen çıkarıldığı bir operasyon yapılmalır. Ameliyatın bir parçası olarak karın bölgesine lokal kemoterapi de uygulanabilir. Ameliyattan sonra bazen radyasyon veya kemoterapi yapılır.
Yaşam beklentisi: DSRCT hastalarının prognozu çok kötüdür, kemoterapi, radyoterapi ve agresif cerrahi rezeksiyona rağmen genel sağkalım yaklaşık % 30 ila % 55 arasındadır [11,12]. Hastaların ancak sadece % 20’sinden daha azı, tanıdan 5 yıl sonra hayatta kalabilmektedir.
4- Sendromik mikroftalmi tip 5
Sendromik mikroftalmi tip 5, anoftalmi*, mikroftalmi** ve retina anormallikleri anormallikleri gibi dizi oküler anomali ile değişken gelişimsel gecikme ve merkezi sinir sistemi malformasyonları karakterize bir genetik hastalıktır. Dünya çapında yaygınlığı yaklaşık <1: 1.000.000 dur. Bugüne kadar dünya çapında 20’den az vaka tanımlanmıştır. Bazı hastalarda ciddi bilişsel gecikme kaydedilirken, bazılarında normal bilişsel gelişim gösterdiği görülmüştür.

Ayrıca büyüme hormonu eksikliğine ve boy kısalığına veya kombine hipofiz hormonu eksikliğine (CPHD) yol açan hipofiz disfonksiyonu da bildirilmiştir.
- Anoftalmi*: Anoftalmi, bebeğin bir veya iki gözü olmadan doğduğu bir doğum kusurudur.
- Mikroftalmi**: Mikroftalmi, doğumdan önce ortaya çıkan bir göz anormalliğidir. Bu durumda, bir veya iki göz küresi anormal derecede küçüktür. Etkilenenlerin bazılarında göz küresi tamamen yokmuş gibi görülse de genellikle bir miktar göz dokusu mevcuttur. Mikroftalmis olan kişilerde ayrıca koloboma*** adı verilen bir durum da olabilir. Koloboma***: Gözü oluşturan yapılardaki eksik doku parçalarıdır. Gözün iris adı verilen kısmında çentikler veya boşluklar da görülebilir.
Sebep: Sendroma, OTX2 genindeki (14q22.3) heterozigot mutasyonlar neden olur [13].
5- Riboz-5-fosfat izomeraz eksikliği
Riboz-5-fosfat izomerazın enzimi eksikliğinden kaynaklanan en nadir görülen genetik bir hastalıktır. Bundan daha nadir görülen bir hastalık yoktur. Beynin beyaz maddesini etkileyen bu hastalık için şu ana kadar dünyada sadece üç vaka rapor edilmiştir.
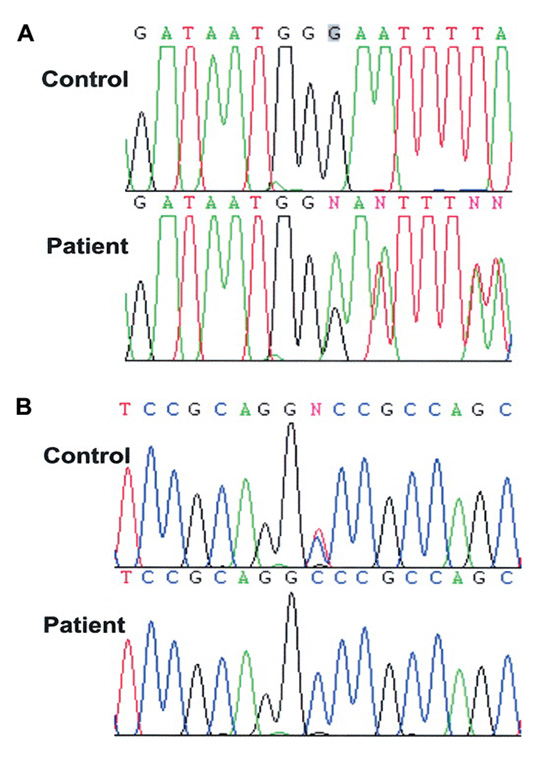
Riboz-5-fosfat izomeraz, glikozu Adenozin Trifosfata (ATP) dönüştürmede rol oynayan önemli bir enzimdir. Bu enzimin eksik olması hastalarda genel gelişimsel gecikme, hareketler ve motor işlevlerde bozulmalara sebep olur.
Klinik Bulgular: Psikomotor gecikme, epilepsi ve ataksi, spastisite, optik atrofi ve sensorimotor nöropati ile çocuklukta başlayan yavaş nörolojik gerilemedir.
Sebep: Riboz-5-fosfat izomeraz eksikliği otozomal çekinik bir hastalıktır. Hastalığa RPIAD geninde meydana gelen mutasyonlar sebep olur. Çocukluk yaşlarında başlar ve dünya çapında yaygınlığı <1/1.000.000 dir [14].
Nadir hastalıklar nasıl tedavi edilebilir
Genetik tedavi: Hızla gelişen genetik tedavi yöntemleri, özellikle nadir hastalıklar için yeni fırsatlar sunmaktadır. Standart uygulamalardan biri, dizayn edilmiş bir DNA dizisinin genetiği değiştirilmiş bir virüs vasıtası ile etkilenen kişinin vücuduna verilmesidir. Virüs hücreye girdikten sonra dizayn edilmiş gen, hücre içine bırakılarak hatalı kısmın tamir edilir.
Örneğin, Spinal musküler atrofiye karşı Zolgensma gen tedavisi 18 Mayıs 2020’de Avrupa İlaç Ajansı (EMA) tarafından onay aldı. Bu tedavi yöntemi ile hastalık tek bir infüzyonla tedavi edilebiliyor. Bir dozu 1.945 milyon Euro olan Zolgensma gen tedavisi dünyada bugüne kadarki en pahalı tedavidir.
İlâçlı tedavi: Avrupa Birliği 2000 yılında nadir hastalıkların ilâçlı tedavisini geliştirilmek ve teşvik etmek için bir yönetmelik çıkardı. Bu yönetmelik ile bu tür ilaçların geliştirmesi için ekonomik teşvikler yaratılmaktadır. Örneğin şiddetli bağışıklık yetmezliği ADA-SCID, metabolik bir hastalık olan alfa-mannosidaz ve bir kas hastalığı olan Duchenne musküler distrofisi’un dahil olduğu 143 nadir hastalık için teşvik almıştır.
Benzer konularda hazırlanmış diğer makaleler
- Teşhisi çok zor, tedavisi çok kolay hastalıklar
- Gen terapi, epilepsi hastaları için yeni bir umut
- Otizmin nasıl başladığına dair yeni ipuçları
- Otizme karşı kanser ilacı?
Dipl. Biologe Mehmet Saltürk
The Institute for Genetics of the University of Cologne
Kaynaklar
- https://www.bundesgesundheitsministerium.de/themen/praevention/gesundheitsgefahren/seltene-erkrankungen.html
- https://www.rarediseaseday.org
- https://www.orpha.net/consor/cgi-bin/Disease_Search_List.php?lng=EN)
- Functional Modeling of the ACVR1 (R206H) Mutation in FOP
- The medical management of fibrodysplasia ossificans progressiva: current treatment considerations
- Insights into the biology of fibrodysplasia ossificans progressiva using patient-derived induced pluripotent stem cells
- Hutchinson–Gilford progeria syndrome: Review of the phenotype
- LMNA mutation in a 45 year old Japanese subject with Hutchinson-Gilford progeria syndrome
- Bestimmung des mutationsspektrums von „desmoplastic small round cell tumors
- Desmoplastic small round-cell tumor: prolonged progression-free survival with aggressive multimodality therapy.
- Results of multimodal treatment for desmoplastic small round cell tumors
- Microphthalmia, Syndromic 5 (MCOPS5)
- Ribose 5-Phosphate Isomerase Deficiency (RPIAD)
YAZIYI PAYLAŞ
Yazar Hakkında
Mehmet Saltuerk
Genetik
Benzer Yazılar
 AIFD Türkiye’de Nadir Hastalıklar Raporu: 5 milyondan fazla kişiyi etkiliyor!
AIFD Türkiye’de Nadir Hastalıklar Raporu: 5 milyondan fazla kişiyi etkiliyor!- Nadir hastalıklardan etkilenen insanların karşılanmamış ihtiyaçlarına odaklanıyoruz
- Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler
- Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri
- ‘Nadir Hastalıklar’ Türkiye’de 7 milyon insanı etkiliyor
 AIFD Türkiye’de Nadir Hastalıklar Raporu: 5 milyondan fazla kişiyi etkiliyor!
AIFD Türkiye’de Nadir Hastalıklar Raporu: 5 milyondan fazla kişiyi etkiliyor! Nadir hastalıklardan etkilenen insanların karşılanmamış ihtiyaçlarına odaklanıyoruz
Nadir hastalıklardan etkilenen insanların karşılanmamış ihtiyaçlarına odaklanıyoruz Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler
Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri
Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri ‘Nadir Hastalıklar’ Türkiye’de 7 milyon insanı etkiliyor
‘Nadir Hastalıklar’ Türkiye’de 7 milyon insanı etkiliyor
YORUMUNUZ VAR MI?