
Kromozomal Hastalıklar – 2: Angelman Sendromu ve Prader-Willi Sendromu
 Angelman Sendromu (AS) ve Prader Willi Sendromu (PWS) 50’li yıllarda tanımlanan ağır genetik iki hastalıktır. Her iki hastalık da toplumda seyrek görülen hastalıklar grubundan olup ikisi de 15. kromozomun 15q11.2-q13 lokasyonundaki hatalardan kaynaklanır. Hastalığa sebep olan bu hata, bazen parçanın (15q11.2-q13) tamamen kopması veya bu bölgedeki bir genin mutasyonu veya 15. kromozom çiftinin değişik kombinasyonundan kaynaklanmaktadır. Her iki hastalığın semptomları doğumla başlar ve ölüme kadar devam eder. Şu anki teknoloji ile her iki hastalığın da geriye dönük tedavisi mümkün değildir.
Angelman Sendromu (AS) ve Prader Willi Sendromu (PWS) 50’li yıllarda tanımlanan ağır genetik iki hastalıktır. Her iki hastalık da toplumda seyrek görülen hastalıklar grubundan olup ikisi de 15. kromozomun 15q11.2-q13 lokasyonundaki hatalardan kaynaklanır. Hastalığa sebep olan bu hata, bazen parçanın (15q11.2-q13) tamamen kopması veya bu bölgedeki bir genin mutasyonu veya 15. kromozom çiftinin değişik kombinasyonundan kaynaklanmaktadır. Her iki hastalığın semptomları doğumla başlar ve ölüme kadar devam eder. Şu anki teknoloji ile her iki hastalığın da geriye dönük tedavisi mümkün değildir.
 Angelman Sendromu (AS) ve Prader Willi Sendromu (PWS) 50’li yıllarda tanımlanan ağır genetik iki hastalıktır. Her iki hastalık da toplumda seyrek görülen hastalıklar grubundan olup ikisi de 15. kromozomun 15q11.2-q13 lokasyonundaki hatalardan kaynaklanır. Hastalığa sebep olan bu hata, bazen parçanın (15q11.2-q13) tamamen kopması veya bu bölgedeki bir genin mutasyonu veya 15. kromozom çiftinin değişik kombinasyonundan kaynaklanmaktadır. Her iki hastalığın semptomları doğumla başlar ve ölüme kadar devam eder. Şu anki teknoloji ile her iki hastalığın da geriye dönük tedavisi mümkün değildir.
Angelman Sendromu (AS) ve Prader Willi Sendromu (PWS) 50’li yıllarda tanımlanan ağır genetik iki hastalıktır. Her iki hastalık da toplumda seyrek görülen hastalıklar grubundan olup ikisi de 15. kromozomun 15q11.2-q13 lokasyonundaki hatalardan kaynaklanır. Hastalığa sebep olan bu hata, bazen parçanın (15q11.2-q13) tamamen kopması veya bu bölgedeki bir genin mutasyonu veya 15. kromozom çiftinin değişik kombinasyonundan kaynaklanmaktadır. Her iki hastalığın semptomları doğumla başlar ve ölüme kadar devam eder. Şu anki teknoloji ile her iki hastalığın da geriye dönük tedavisi mümkün değildir.Aşağıda ayrıntılarını okuyacağınız iki hastalığın her ne kadar genetik sebepleri benzerlik gösterse de etkileri oldukça farklıdır. Hastalıklar hakkında ayrıntıya geçmeden önce konunun daha iyi anlaşılması için makalede oldukça fazla geçen gen, kromozom ve delesyon hakkında birkaç temel bilgi vermek kanımca yerinde olacaktır.
Gen nedir?
Genler, anne ve babamızdan aldığımız ve çocuklarımıza aktardığımız, hayatın başlangıcından sonuna kadar bizi biz eden kalıtım birimidir. Genler, Adenin (A), guanin (G), sitozin (C), timin (T) olmak üzere 4 farklı yapı taşının değişik kombinasyonlarda yan yana dizilmesi ile meydan gelirler. Bu yapı taşlarına Nükleotid denir.
Kromozomal hastalıklar (Trizomi 21-18-13) ve Down sendromu, tanı ve tedavisi
Vücudumuzun her hücresinden yaklaşık 3 milyar Nükleotid vardır ve genler bu 3 milyar Nükleotid’in belirli bölgelerinde yer almaktadır. Tam kesin sayı bilinmemekle birlikte insanda yaklaşık 20.000 ile 30.000 arası gen bulunmaktadır (link).
Bu genlerin büyüklükleri ve işlevleri birbirinden çok farklıdır ve genlerin düzenli çalışması ile sağlık arasında doğrudan ilişki vardır. Genlerin sadece sağlığımızla değil aynı zamandan bizim fiziksel görünümüzle de doğrudan ilgisi vardır. Örneğin göz rengi, saç rengi, cild rengi hatta parmaklarımızın uzunluğuna varana kadar birçok irili ufaklı ayrıntı ve bizi biz yapan sayısız özellik genlerimiz tarafından belirlenir….
Ama genlerin görevi sadece bunlarla da kalmaz. Genlerimiz aynı zamanda bizim karakterimizin şekillenmesinde de rol oynarlar. Örneğin tutkular, sevgiler, nefretler, korkular, alışkanlıklar gibi yaşama dair her şey genlerimiz tarafından şekillenir. Yani genlerimiz bizim kaderimizdir.
Kromozom nedir
Kromozomlar, üzerinde genlerin bulunduğu makro kalıtım birimleridir. Her insan anneden 23, babadan 23 gelmek üzere 46 adet kromozoma sahiptir. Yukarıda bahsedilen 3 milyar nükleotid bu 23 çift kromozom üzerinde adeta paketlenmiş bir şekilde yer alır. Her kromozomdaki nükleotid sayısı ve buna bağlı olarak gen sayısı farklı farklıdır.
Örneğin 1 numaralı kromozom, en büyük kromozomdur ve üzerinde 247 milyon nükleotid ve 3148 gen bulunur. Kromozom numarası büyüdükçe geninin fiziksel boyutu küçülür ve üzerindeki nükleotid sayısı da azalır. Ama nükleotid sayısının azalması daha az sayıda gen anlamına gelmez. Örneğin 191 milyon nükleotid bulunan 4 kromozomda 453 gen bulunurken, 132 milyon nükleotid bulunan 12. kromozomda 1330 gen bulunur.
Yeri gelmişken birazda kromozomların yapısından da kısaca bahsetmekte fayda var. Kromozomlar bir uzun bir de kısa olmak üzere iki koldan oluşur. Yukarıdaki kısa kola „p“, aşağıdaki uzun kola „q“ kolu denir. Kromozomlar üzerinde her biri kendine özgü bantlardan oluşur. Kromozomlar bu bantlaşma örüntüsüne dayanarak teşhis edilirler.
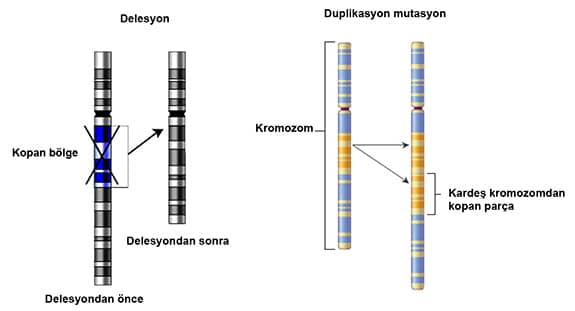
Genlerin kromozom üzerindeki lokasyonu bu bandaların numarasına göre yapılır. Örneğin, 15q11.2-q13 lokasyonu şu anlama gelmektedir: 15. kromozomun uzun kolunun 11.2 bandından 13´e kadar olan bölgedir.
Delesyon nedir?
Delesyon genetikte bir kromozomun bir parçasının kopması veya, kaybolmasıyla meydana gelen kromozom anomalileri olarak tanımlanır. Kopan parça bazen küçük olabileceği gibi bazen büyükte olabilmektedir. Kopan parça üzerinden birçok gen olduğu için kopan parçayla birlikte genler de kaybolur.
Genlerin kaybolması ile birlikte bu genlerin yerine getirdiği birçok görev yapılamaz olur ve Angelman Sendromu (AS) ve Prader Willi Sendromu(PWS) gibi birçok hastalık ortaya çıkar. Bu hastalıkların şiddeti kopan parçanın büyüklüğüne göre farklılık gösterir. Bazen de kopan parça kardeş kromozoma yapışarak o kromozomun gen dengesinin bozulmasına sebep olur.
Angelman-Sendromu (AS)
Angelman sendromu, doğumla birlikte ortaya çıkan, fiziksel ve zihinsel gelişimi geciktiren, nadir görülen kalıtsal bir bozukluktur. Hastalık ilk olarak 1965 yılında İngiliz çocuk doktoru Harry Angelman tarafından tanımlandı. Sendromun ortalama insidens 1:15.000 ila 1:20.000 arasındadır.
Angelman sendromu belirtiler
Belirtilerin yoğunluğu hastanın durumuna göre değişmekle birlikte, hastaların yüzlerinde sürekli gülümseme ifadesi ve ortalamadan daha fazla gülmeleri ile dikkat çekerler. Bu yüzden hastalık ingilizce konuşulan ülkelerde ‘Happy Puppet Sendromu’ (Mutlu bebek sendromu) olarak da adlandırılır. Angelman sendromluların dikkat çeken başka bir karakteristik özelliği ise yürürken denge sorunlarının olmasıdır.
Yukarıda belirtilen ana semptomlara ek olarak, bu hastalarda birçok semptom daha bulunur. Örneğin sıklıkla görülen epileptik nöbetler, üç yaşından itibaren fark edilmeye başlayan küçük bir baş (mikrocefaly olarak adlandırılır) ve beynin Elektroensefalografi (EEG ) çekimlerinde elektriksel aktivitelerde anormallikler…
Angelman sendromunda da yaygın görülen başlıca semptomlar şöyle özetleyebiliriz
- Hiperaktivite, huzursuzluk
- Az ya da çok hareket veya denge bozuklukları/Ataksi (Genellikle koordinasyonsuz hareketler)
- Konuşma zorluğu. Genellikle konuşmayı anlar ama konuşma yapmadan jestlerle iletişim kurar
- Hafif bir heyecanlanma hali, çoğunlukla sallanma
- Salya akması ve aşırı çiğneme hareketleri
- Uyku-uyanıklık ritimtinde bozukluk ve az uyku gereksinimi
- Kâğıt , su ve plastikten çıkan sese büyük ilgi gösterme
- Beslenme alışkanlığında farklılıklar (Örneğin büyük iştahla yemek yemek, ya da yenmeyen şeyleri yeme)
- Küçük bir kafa (mikrosefali) ve beyin dalgalarındaki düzensizlikler
- Kafa arkasıda düzlük
- Büyük çene ve geniş ağız
- Geniş aralıklı dişler
- Uzanan dil
- Şaşılık
- Az pigmentli cilt, açık saç ve göz rengi
- Bebeklikte beslenme sorunları
- Düz tabanlık
- Isı hassasiyeti
- Çocukluk döneminde kilolu olma
- Omurganın eğriliği (skolyoz)
- Hipermotor davranış (hareket artışı, huzursuzluk)
- Epileptik nöbetler
- Görme bozuklukları.
- Gelişimsel gecikme
Angelman sendromu teşhisi
Angelman sendromunun belirtileri diğer rahatsızlıklarla benzerlik göstermesi nedeni ile gerek doğum öncesi hamilelik döneminde, gerekse doğum sonrası bebeklik ve erken çocukluk döneminde pek fazla fark edilmeyebilir. Bu nedenle hastalığa ilk tanı genellikle 3 ve 7 yaşları arasında konur.
Angelman sendromunun genetik sebepleri
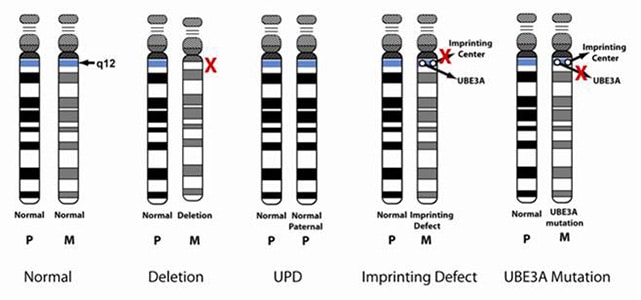
Angelman sendromu, 15. Kromozomdaki bir anormalliğe bağlı olarak ortya çıkan kalıtsal bir hastalıktır. Hastalık, genellikle anneden alınan 15. Kromozomun küçük bir parçasının eksik olması (mikro-delesyon) nedeniyle ortaya çıkar.
Vakaların % 10‘u yumurta döllendikten sonra hücre bölünmesi sırasında meydana gelen mutasyondan kaynaklanır. Bu tür vakalarda annenin 15. Kromozomunda hatalı veya eksik bir kısım bulunmamaktadır. Yani genetik kusur(mutasyon) yumurta döllendikten sonra hücresinin gelişmesi esnasında ortaya çıkar. Çok nadir de olsa babanın 15. Kromozomundan kaynaklanan Angelman sendromu vakaları da görülmektedir.
Yaklaşık Angelman sendromlu her 10 çocuğun birinde 15. kromozom çiftinde herhangi bir anormallik yoktur. Böyle durumlar genetik değişikliğin çok küçük olduğu durumlarda görülür ki, bu vakalar bile artık günümüzde mevcut tekniklerle tespit edilebilmektedir.
Angelman sendromu çeşitli genetik hatalardan kaynaklanır. Bunlar sırasıyla şöyledir
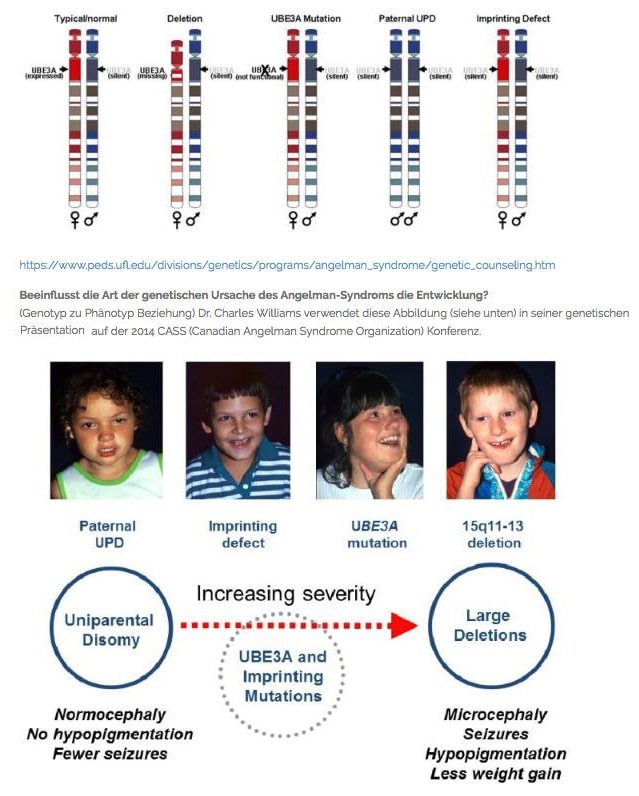
- Mikrodelesyon 15q11.2-q13: Anneden gelen 15. kromozom üzerinde küçük bir parçanın(15q11.2-q13) eksik olmasından kaynaklanır (bu mikrodeletiyon olarak adlandırılır). Bu tür vakalar tüm Angelmann sendromu vakalarının %70 oluşturur.
- Paternal Uniparentale Disomie 15 (UPD): Koromozom çiftinin her ikisi de babadan gelir. Anneden gelen 15. kromozom çocukta bulunmaz. Bu tür Angelmann sendromu vakaları tüm vakaların %1 ni oluşturur.
- Imprinting center mutations: 15 Kromozomun her ikisinde de Imprinting centerde çalışmaz. 15 Kromozomda babaya ait küçük bir bölgenin annenin 15 kromozomunda yer almasından kaynaklanır. Görülme sıklığı % 4 dir.
- Mutation in UBE3A-Gen: 15 Kromozom(anneden) üzerindeki UBE3A-Geninde meydana gelen mutasyondan kaynaklanır. Angelmann sendromu vakalarının % 5-10 nu oluşturur.
Angelman sendromu tedavisi
Angelman sendromu, genomda meydana gelen bir anormallikten kaynaklandığı için genlerde geriye dönük bir düzenleme şimdilik mümkün değil. Ancak özenli bakım, sevgi ve yoğun pedagojik yardım ile semptomları hafifletmek ve hastanın yaşamını kolaylaştırmak, yaşam kalitesini yükseltmek mümkün. Her ne kadar şu anki teknoloji ile hastalığı tamamıyla iyileştirmek mümkün olmasa da gen teknolojisindeki gelişmeler birçok hastalıkta olduğu gibi Angelmann sendromuda da umutları taze tutuyor.
Angelman sendromunun güncel tedavi metotları
- Ergoterapi: Hareket terapisi, fiziksel ya da zihinsel etkinlikler yapılarak çocukların mümkün olduğu kadar günlük yaşama katılmaları sağlanır.
- Konuşma terapisi: Konuşma ve konuşma bozukluklarının tedavisi
- Mototerapi: Hareket ve davranıştaki hatalar ve eksiklikler düzeltilir
- Hipoterapi: At eşliğinde terapi yapılarak fiziksel, zihinsel veya duysal iyileşme ve gelişmesi sağlanır.

Prader-Willi Sendromu (PWS) nedir?
Prader-Willi sendromu da Angelman sendromu gibi nadir görülen genetik bir hastalıktır. Hastalık ilk olarak 1950’li yıllarda Prader ve Willi adında iki çocuk doktoru tarafından tanımlandı ama hastalığın nedenleri ancak 1981’de çözülebildi.
Dünya genelinde yaklaşık 350.000 – 400.000 kişinin Prader-Willi sendromu olduğu tahmin ediliyor. PWS`nin toplumda görülme sıklığı yaklaşık 1:10,000 ila 1:15.000 dir ve kadın ve erkekte dağılımı aşağı yukarı eşit orandadır.
Prader Willi Sendromu (PWS) Belirtiler
Prader-Willi sendromunun belirtileri hamilelik döneminde pek belirgin olmadığı için bu hastalık da Angelmam sendromu gibi pek fark edilmeyebilir.
- Hastalığın ilk belirtileri bebeklik döneminde kendini göstermeye başlar. Bu dönemde genellikle beslenme güçlüğü (içmede problemler) ve kaslarda zayıflık görülür.
- Bebeklik döneminde göze çarpan başka bir özellik ise genital organlarda görülür. Genital organlar eksik veya gözle görülür derecede küçüktür (genital hipoplaziler).
- Yaş ilerledikçe çocuğun yemek yeme davranışında değişimler başlar, daha fazla yemeye ve doygunluk hissini hissetmemeye başlarlar. Bu nedenle Prader-Willi sendromlu çocukların en karakteristik özelliklerinden biri de obez olmalarıdır.
- Vücut çok az büyüme hormonu oluşturduğu için küçük bir vücut yapısına sahiptirler
- Küçük eller ve ayaklar göze çarpan başka bir özelliktir(Akromikri)
- Geç çocukluk ve ergenlik döneminde kasıklarda kıllanma gibi bazı belirtileri görülebilir ancak diğer fiziksel gelişim belirtileri ya gecikmiştir veya hiç görülmez
- Ergenlikte cinsel organlarda az veya eksik gelişme. Testisler testisler torbasına inmemiştir(kriptorşidizm).
- Kızlarda, ilk menstruasyon çok geç gerçekleşir.
- Cinsiyet hormonlarının yeterli olmamasına bağlı olarak kısırlık ve kemiklerde kırılganlık (osteoporoz).
- İlerleyen yaşlarda çoğunlukla tip 2 diyabet, tiroid bezlerinde işlevsel bozukluk veya uyku apnesi görülebilir.
- Omurgada deformasyon(skolyoz).
- Birçok hastada gözlerini kırpıştırma.
- Çoğunlukla, zihinsel gelişim yavaş ve konuşulanı anlamada zorluklar.
- Bazı davranışlar otizmle oldukça benzerlik gösterir.
Prader Willi Sendromu (PWS) Teşhis
Yeni doğan bebeklerde Prader Willi sendromu teşhisi ancak oluşan semptomlara dayanarak yapılabilir. Örneğin, ön teşhis için açıklanamayan zayıflık, yeme ve içmede zorluk gibi semptomlar bazen yeterli olabilmekle birlikte bazen bu belirtiler doktorun ve ailenin dikkat çekmeye bilir.
Ayrıca çocukluk ve ergenlik çağında kandaki büyüme hormonu ile cinsel hormonların (östrojen, testosteron, FSH, LH) ölçümü de Prader-Willi-Syndromu teşhis için bir fikir verebilir. Beyin dalgalarının incelenmesi de(elektroensefalografi, EEG) Prader-Willi- Sendromu teşhisine yardımcı olan bir başka teknikdir. Tabii ki kesin karar gen testi yapılarak verilir.
Prader Willi-Sendromu çeşitli genetik hatalardan kaynaklanır. Bunlar sırasıyla şöyledir
1- Mikrodeletion 15q11.2-q13: Tıpkı Angelmann sendromunda olduğu gibi Prader-Willi- Sendromunda da 15. Kromozomda küçük bir parçanın (15q11.2-q13) kaybolması ile ortaya çıkar. Angelman sendromundan tek fark kaybolon parçanın babadan alınan 15. kromzomda gerçekleşmesi. Bunlar tüm Prader-Willi- Sendromu vakalarının %70’ni oluşturur.
(Not: Kopan parça yüksek çözünürlüklü, mikroskopla tespit edilebiliyor. Ayrıca çok az hastada 15. kromozomun SNORD116 adındaki bir başka bölgesinde(lokus) olağandışı küçük bir silinmeden kaynaklanan Prader-Willi- Sendromu vakaları da bulunmaktadır.)
2- Maternale Uniparentale Disomie 15 (UPD): Bu hata, bir adet anneden, bir adet de babadan gelmesi gereken gereken 15. krorozomun genetik bir hatadan dolayı her ikisinin de anneden gelemesinden kaynaklanı Yani Maternale Uniparentale Disomie de 15. kromozomun her ikisi de annneden gelmiştir. Bu tür Prader-Willi- Sendromu vakaları tüm vakaların % 25 ila %30 nu oluşturur.
3- Imprinting center mutations: 15 Kromozomun her ikisinde de Imprinting centerde çalışmaz. 15 kromozomda anneye ait küçük bir bölgenin babanın 15 kromozomunda yer almasından kaynaklanır. Prader-Willi sendromunun nadir bir formudur. PWS vakalarının sadece % 1’i bu hatadan kaynaklanır.
Prader Willi Sendromu Tedavisi
Prader-Willi sendromu da genomda meydana gelen bir anormallikten kaynaklandığı için geriye dönük bir düzenleme şimdilik mümkün değildir. Tedavi daha çok semptomları hafifletmeye yöneliktir. Örneğin, beslenme kontrolü, fizyoterapi, düzenli egzersiz ve psikolojik ve eğitim desteği uzun vadede bu olumlu sonuçlar vermektedir. Aşırı kiloyu azaltma ve hormonları düzenleme, davranış sorunlarını ilaçla (örn., Nöroleptikler) düzenleme gibi önlemlerle hastanın yaşam kalitesi yükseltilir.
Hastalığın derecesi ve kişiden kişiye değişen bireysel şekillenmeleri hafifletmek için ne gibi bireysel tedavi yöntemleri uygulanabileceği henüz tam olarak bilinmiyor. Bununla birlikte, klinik çalışmalar büyüme hormonu tedavisi ile boy uzunluğunun normalleştirebileceği ve vücut yağ seviyelerini düşürebileceğini gösteriyor.
Benzer konuda hazırlanmış diğer makaleler
- Kromozomal hastalıklar-1 (Trizomi 21, Trizomi 18 ve Trizomi 13)
- Kromozomal bozukluklar ve cinsiyet belirsizliği
- Genler ve hastalıklar
- Hatalı gen intihar riskini arttırıyor
Mehmet Saltuerk
++++++++++++++++++++++++
Dipl. Biologe Mehmet Saltürk
The Institute for Genetics
of the University of Cologne
++++++++++++++++++++++++
Kaynaklar
The Human genome project (HGP)
Angelman Syndrome
Das Prader-Labhart-Willi-Syndrom im Säuglingsalter
Highly restricted delesyon of the SNORD116 region is implicated in Prader–Willi Syndrome
UBE3A/E6-AP mutations cause Angelman syndrome
Prader-Willi, Angelman, and 15q11-q13 duplication syndromes
Angelman's syndrome and 15q11-q13 delesyon.
Prader-Willi syndrome and a delesyon/duplication within the 15q11-q13 region
Prader-Willi and Angelman Syndromes: Disorders of Genomic Imprinting
Bu blogdaki makaleler bir başka yayın organında kaynak gösterilmeden yayınlanamaz, çoğaltılamaz ve kullanılamaz.
YAZIYI PAYLAŞ
Yazar Hakkında
Mehmet Saltuerk
Genetik
Benzer Yazılar
 Kromozomal hastalıklar (Trizomi 21-18-13) ve Down sendromu, tanı ve tedavisi
Kromozomal hastalıklar (Trizomi 21-18-13) ve Down sendromu, tanı ve tedavisi- Kromozomal hastalıklar ve gebelik: 35 yaş üstü hamilelikler neden riskli
- Down sendromu nedir, neden olur? Belirtileri ve tedavisi
- Genetik hastalıklar: Dravet sendromu nedir? Nedenleri ve tedavisi
- Turner Sendromu nedir? Belirtileri, nedenleri ve tedavisi
 Kromozomal hastalıklar (Trizomi 21-18-13) ve Down sendromu, tanı ve tedavisi
Kromozomal hastalıklar (Trizomi 21-18-13) ve Down sendromu, tanı ve tedavisi Kromozomal hastalıklar ve gebelik: 35 yaş üstü hamilelikler neden riskli
Kromozomal hastalıklar ve gebelik: 35 yaş üstü hamilelikler neden riskli Down sendromu nedir, neden olur? Belirtileri ve tedavisi
Down sendromu nedir, neden olur? Belirtileri ve tedavisi Genetik hastalıklar: Dravet sendromu nedir? Nedenleri ve tedavisi
Genetik hastalıklar: Dravet sendromu nedir? Nedenleri ve tedavisi Turner Sendromu nedir? Belirtileri, nedenleri ve tedavisi
Turner Sendromu nedir? Belirtileri, nedenleri ve tedavisi
YORUMUNUZ VAR MI?