
Kök hücre motor nöron hastalığına çare olabilir

Son kök hücre çalışmaları, motor nöron hastalıklarında (MND) yeni tedavi arayışını hızlandırdı. Proceedings of the National Academy of Sciences dergisinin yeni sayısında yayımlanan bir çalışmaya göre, kalıtımsal MND olan bir hastanın cilt hücreleri kullanarak, motor nöronlar üretilebildi. Edinburgh Üniversitesi Euan MacDonald MND Araştırma Merkezi Direktörü Prof. Dr. Siddharthan Chandran, hasta kök hücrelerinin model MND için kullanılmasının, bu hastalığın nedeninin öğrenilmesi ve maliyet etkin test yöntemleri ile yeni ilaçların geliştirilmesi olasılığını artırdığını belirtti.
Çalışmada MND’lerin %90’undan fazlasında TDO-43 adı verilen protein anormallikleri keşfedildi. İlk kez bilim adamları anormal TDP-43’ün insan motor nöronlar üzerindeki doğrudan etkisini görebildi. Bu gelişme, MND’li hastalarda hücresel olayları ortaya çıkaran bir laboratuvar modeli oluşturmak için önemli bir yapı taşı.
Körlük ve kök hücre tedavisi: Yeni geliştirilen yöntem körlüğe çare olabilir
Aynı zamanda MND Derneği Araştırma ve Geliştirme Direktörü Dr. Brian Dickie’ye göre, tüm dünyada bu hastalığı anlamak ve yenmeye odaklanmış önemli kurumlardan önemli isimlerin katıldığı uluslararası işbirliğinin de önemini ortaya koyar nitelikte. MND hareketi, konuşmayı ve solunumu kontrol eden motor sinirlerin ilerleyici kaybından kaynaklanan yıkıcı, tedavisi olmayan ve son derece ölümcül bir hastalık.
MND Derneği’nin finansman sağladığı çalışma, Proceedings of the National Academy of Sciences’da yayımlandı. Edinburgh Üniversitesi Euan MacDonald MND Araştırma Merkezi tarafından yapılan çalışmada King’s College London, Columbia, New York ve San Francisco Üniversiteleri de yer aldı.
KAYNAK: Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. B. Bilican, A. Serio, S. J. Barmada, A. L. Nishimura, G. J. Sullivan, M. Carrasco, H. P. Phatnani, C. A. Puddifoot, D. Story, J. Fletcher, I.-H. Park, B. A. Friedman, G. Q. Daley, D. J. A. Wyllie, G. E. Hardingham, I. Wilmut, S. Finkbeiner, T. Maniatis, C. E. Shaw, S. Chandran. Proceedings of the National Academy of Sciences, 2012; DOI: 10.1073/pnas.1202922109
Makalenin tam metnine aşağıdaki linkten ulaşılabilmektedir
Abstract: Transactive response DNA-binding (TDP-43) protein is the dominant disease protein in amyotrophic lateral sclerosis (ALS) and a subgroup of frontotemporal lobar degeneration (FTLD-TDP). Identification of mutations in the gene encoding TDP-43 (TARDBP) in familial ALS confirms a mechanistic link between misaccumulation of TDP-43 and neurodegeneration and provides an opportunity to study TDP-43 proteinopathies in human neurons generated from patient fibroblasts by using induced pluripotent stem cells (iPSCs).
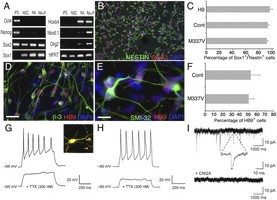
Here, we report the generation of iPSCs that carry the TDP-43 M337V mutation and their differentiation into neurons and functional motor neurons. Mutant neurons had elevated levels of soluble and detergent-resistant TDP-43 protein, decreased survival in longitudinal studies, and increased vulnerability to antagonism of the PI3K pathway. We conclude that expression of physiological levels of TDP-43 in human neurons is sufficient to reveal a mutation-specific cell-autonomous phenotype and strongly supports this approach for the study of disease mechanisms and for drug screening.
YAZIYI PAYLAŞ
Benzer Yazılar
 Kök hücre nedir? Kök hücre tedavisi ne işe yarar? Nakli nasıl yapılır?
Kök hücre nedir? Kök hücre tedavisi ne işe yarar? Nakli nasıl yapılır?- Körlük ve kök hücre tedavisi: Yeni geliştirilen yöntem körlüğe çare olabilir
- Kök hücre tedavisi kıkırdak ve menisküs yaralanmalarına çözüm oluyor
- Biyolojide devrim! CRISPR/Co9 teknolojisi ile cilt hücreleri kök hücreye dönüştürüldü
- Kök hücre tedavisi ile gençleşmek mümkün mü, yaşlılık hastalıklarına çare mi?
 Kök hücre nedir? Kök hücre tedavisi ne işe yarar? Nakli nasıl yapılır?
Kök hücre nedir? Kök hücre tedavisi ne işe yarar? Nakli nasıl yapılır? Körlük ve kök hücre tedavisi: Yeni geliştirilen yöntem körlüğe çare olabilir
Körlük ve kök hücre tedavisi: Yeni geliştirilen yöntem körlüğe çare olabilir Kök hücre tedavisi kıkırdak ve menisküs yaralanmalarına çözüm oluyor
Kök hücre tedavisi kıkırdak ve menisküs yaralanmalarına çözüm oluyor Kök hücre tedavisi ile gençleşmek mümkün mü, yaşlılık hastalıklarına çare mi?
Kök hücre tedavisi ile gençleşmek mümkün mü, yaşlılık hastalıklarına çare mi?
YORUMUNUZ VAR MI?