
Fenilketonüri (PKU) nedir? Nedenleri, belirtileri, tedavisi ve diyeti

Fenilketonüri, (PKU) genetik (ırsi) bir hastalıktır ve genellikle akraba evliliklerinde görülür. Fenilalanini adlı amino asidi parçalamak için gereken enzimi ürettiren gendeki kusurdan kaynaklanır. Enzim görevini yerine getirmediği için, alınan fenilalanin kanda yükselir. Bu nedenle beyine ciddi ve kalıcı hasarlar oluşabilir. Bir kişide PKU hastalığı gelişmesi için, hem anne hem de babanın, kusurlu gene sahip olması gerekir. Fenilketonüri hastaları protein içeren yiyecekler veya aspartam tükettiğinde ciddi sağlık sorunları yaşarlar. PKU hastalarının hayat boyu, protein içeren gıdaları sınırlı tüketmelidir. Doğum sonrasında bebeklerde fenilketonüri taraması yapmak, hastalığın erken teşhis edilmesini ve bebeklerde zihinsel kusur olmasını engellemek için hayati öneme sahiptir.
Table of Contents
Fenilketonüri (PKU) nedir?
Fenilketonüri, vücutta fenilalanin adı verilen bir amino aside neden olan nadir bir genetik durumdur. Amino asitler, proteinin yapı taşlarıdır. Fenilalanin, tüm proteinlerde ve bazı yapay tatlandırıcılarda bulunur. Kanda aşırı fenilalanin birikimi, merkezi sinir sisteminde hasara neden olabilir. Yenidoğan bebeklerde tespit edilip tedavi edilmezse, hastalık kalıcı beyin hasarına neden olabilir. Nörolojik ve fiziksel bozulmayı önlemek için yaşam boyu tedavi gereklidir.
Fenilketonüri (PKU) nedenleri
Fenilketonüri, PAH genindeki bir kusurun neden olduğu kalıtsal bir durumdur. Fenilketonüri hastasında, bu kusurlu gen, bir amino asit olan fenilalanini işlemek için gereken enzimin eksikliğine neden olur. PKU hastaları süt, peynir, fındık ve et gibi protein açısından zengin besinler, ekmek ve makarna gibi tahıllı gıdalar veya yapay tatlandırıcı aspartam tükettiğinde vücutlarında tehlikeli bir fenilalanin birikimi başlar. Bu fenilalanin birikimi, beyindeki sinir hücrelerine zarar verir.
Bir çocuğun PKU hastalığına yakalanması için, hem anne hem de babanın, kusurlu gene sahip olması gerekir. Yalnızca bir ebeveynde bu kusurlu gen varsa, çocukta hiçbir belirtisi görülmez, ancak bu genin taşıyıcısı olur.
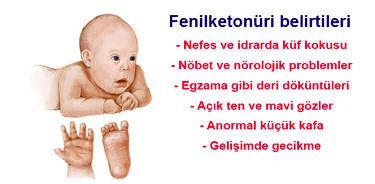
Fenilketonüri (PKU) belirtileri
- Nöbetler ve nörolojik problemler
- Egzama gibi deri döküntüleri
- Açık renk ten ve mavi gözler
- Kişinin nefesinde, deri ve idrarında küf kokusu
- Anormal derecede küçük kafa (mikrosefali)
- Hiperaktivite
- Gelişimde gecikme
- Psikolojik bozukluklar
Fenilketonüri belirtileri hafif ile şiddetli arasında değişebilir. Bu bozukluğun en şiddetli formu klasik Fenilketonüri olarak bilinir. Klasik PKU hastası bir bebek hayatının ilk birkaç ayında normal görünebilir. PKU doğumda teşhis edilmezse ve tedavi hızlı bir şekilde başlatılmazsa, bozukluk aşağıdakilere neden olabilir:
- Hayatın ilk birkaç ayı içinde geri dönüşümsüz beyin hasarı ve zihinsel engeller
- Daha büyük çocuklarda davranış problemleri ve nöbetler
Daha az ciddi bir fenilketonüri formu olan varyant FKÜ’de bebeklerde sadece hafif belirtiler olabilir, ancak zihinsel engeli önlemek için özel bir diyet izlemeleri gerekir. Belirli bir diyet ve diğer gerekli tedaviler başladıktan sonra, belirtiler azalmaya başlar. Diyetlerini doğru şekilde yöneten FKÜ hastaları genellikle herhangi bir semptom göstermez.
Fenilketonüri (PKU) testi
Yenidoğan kan testi, neredeyse tüm fenilketonüri vakalarını teşhis etmek için yeterlidir. Eğer fenilketonüri hastası iseniz veya aile öykünüz varsa, gebelik veya doğum öncesi tarama testlerini yaptırmanız gerekebilir. PKU taşıyıcıları bir kan testi ile teşhis edilebilir.
Tarama testi, bebek bir ila iki günlükken ve hala hastanede iken topuktan alınan kan ile gerçekleştirilir. Bir laboratuvar, PKU dahil olmak üzere belirli metabolik bozukluklar için kan örneğini test eder. Bu test bebeğinizin fenilketonüri hastalığına sahip olabileceğini gösteriyorsa, tanıyı doğrulamak için daha fazla kan ve idrar testi de dahil olmak üzere ek testler gerekebilir. Bu testler PKU hastalığına neden olan PAH gen mutasyonunun varlığını araştırır. Bu testler genellikle doğumdan altı hafta sonra yapılır.
Bir çocuk ya da yetişkin örneğin gelişimsel gecikmeler gibi PKU belirtileri gösteriyorsa, tanıyı doğrulamak için yine bir kan testi gerekebilir. Hastadan kan örneği alınır ve fenilalanini parçalamak için gereken enzimin varlığı analiz edilir.
Fenilketonüri değerleri
Fenilketonüri test sonucunda kanda bulunan fenilalanin düzeyinin:
- 0-12 yaş arası 2-6 mg/dl
- 12 yaşından sonra 2-11 mg/dl değerleri arasında bulunması normal kabul edilir.
Değerler bu düzeylerin üzerindeyse PKU hastalığından şüphelenilebilir.
Fenilketonüri (PKU) tedavisi
Fenilketonüri hastaları, özel bir diyet ve ilaç tedavisiyle belirtilerini kontrol altına alabilir ve komplikasyonları önleyebilirler.
Fenilketonüri diyeti
Fenilketonüri, tedavisinin ana yolu, fenilalanin içeren gıdaları sınırlayan özel bir diyet uygulamaktır. PKU hastası bebekler anne sütü ile beslenebilir. Genellikle Lofenalac olarak bilinen özel bir mama tüketmeleri gerekir. Bebeğiniz katı yiyecekleri yiyebilecek kadar büyüdüğünde, protein açısından zengin yiyecekler yememelidirler.
Protein nedir? Hangi besinlerde bulunur? Faydaları ve zararları
PKU hastaları hangi yiyecekleri yememeli
- Yumurta
- Et ve et ürünleri (tavuk, balık, hindi, kırmızı et, salam, sosis, sucuk, sakatat vb…)
- Süt ve süt ürünleri (yoğurt, ayran, cacık, peynir)
- Kuruyemişler
- Kuru fasulye, mercimek, nohut, bakla, soya çeşitleri, barbunya
- Makarna, ekmek, poğaça, yumurta, simit, kraker, bisküvi
- Aspartam içeren içecekler
Fenilketonüri formülü
Yeterli miktarda protein aldıklarından emin olmak için Fenilketonüri hastası çocukların PKU formülünü tüketmeleri gerekmektedir. Özel bir besin takviyesi olan PKU formülü, Fenilalanin dışında vücudun ihtiyaç duyduğu tüm amino asitleri içerir. Ayrıca özel sağlık mağazalarında bulunabilen düşük proteinli, PKU dostu gıdalar da vardır. Her insan için güvenli olan fenilalanin miktarı farklıdır ve zamanla değişebilir. Doktorunuz aşağıdakiler aracılığıyla güvenli bir miktar belirleyebilir:
- Diyetinin, büyüme çizelgesinin ve fenilalanin kan düzeyinin düzenli gözden geçirilmesi
- Özellikle çocukluk döneminde büyüme ve gebelik sırasında değişkenlik gösterebilen
- Fenilalanin düzeylerini izlemeye yarayan kan testleri
- Büyüme, gelişme ve sağlığı değerlendiren diğer testler
Ayrıca Fenilketonüri diyetini öğrenmenize yardımcı olacak, gerektiğinde diyetinizde ayarlamalar yapabileceğiniz ve PKU diyet zorluklarını yönetmenin yolları konusunda öneride bulunabilecek uzman bir diyetisyene danışmalısınız.
Çocuklarda bağışıklık sistemini güçlendirmek için 4 altın kural
Nötr amino asit tedavisi
Fenilketonüri diyetine toz veya tablet formunda nötr amino asit terapisi isimlendirilen bir besin takviyesi de eklenebilir. Bu destek, fenilalaninin emilimini engelleyebilir. PKU hastası olan yetişkinler bu takviyeyi doktorlarına veya diyetisyenlerine danışmalı.
Fenilketonüri ilaç tedavisi
ABD Gıda ve İlaç Dairesi (FDA), Fenilketonüri tedavisi için sapropterin (Kuvan) kullanımını onayladı. Sapropterin, fenilalanin düzeyinin düşürülmesine yardımcı olur. Bu ilaç, özel bir PKU diyeti ile birlikte kullanılmalıdır. Ancak, bütün Fenilketonüri hastalarında işe yaramayabilir. Hafif fenilketonüri vakaları olan çocuklarda daha etkilidir.
Fenilketonüri hastalarına öneriler
- Hastalığınızı tanıyın. PKU ile ilgili gerçekleri bilmek, durumdan sorumlu olmanıza yardımcı olabilir.
- Aynı sorunu yaşayan diğer ailelerle tanışın. Bunun için yerel veya çevrimiçi destek gruplarından yararlanabilirsiniz.
- Diyet planlamanız için bu alanda uzman bir diyetisyenden yardım alın.
- Ev dışında da yemek yeme planları yapın. Bir restoranda yemek yemek tüm aile için eğlenceli olabilir.
- Mali yardım kaynakları bulun. Besin takviyeleri ve düşük proteinli gıdaların yüksek maliyetlerini karşılamaya yardımcı olacak programlar veya sigorta planları varsa öğrenmeye çalışın.
- PKU hastası bir çocuğunuz varsa onu spor, müzik ya da sevdiği hobilere odaklanmaya teşvik edin, Böylece yiyemedikleri yiyecekler yüzünden daha az moralleri bozulacaktır.
- Çocuğunuzun diyetini mümkün olduğunca erken öğrenmesine ve uygulamasına yardım edin.
- Evde herkesin yiyebileceği yemeklere ağırlık verin.
- Eğer piknik ve araba gezileri yapacaksanız önceden plan yapın ve PKU dostu yemek seçeneklerini değerlendirin.
- Çocuğunuzun okulundaki öğretmenler ve diğer personel ile konuşun. Eğer önemini ve nasıl işlediğini anlatırsanız, çocuğunuzun öğretmenleri ve kafeterya personeli, PKU diyetiyle ilgili çok yardımcı olabilirler.
- Olumlu ve problem çözücü bir gıda tavrı sürdürün. Bknz:>>> Phenylketonuria
YAZIYI PAYLAŞ
Benzer Yazılar
 Her 100 kişiden 4’ünde görülen PKU hastalığına karşı farkındalık çok önemli
Her 100 kişiden 4’ünde görülen PKU hastalığına karşı farkındalık çok önemli PKU Hasta Araştırması: Fenilketonüri hastaları ‘yorgun, üzüntülü ve endişeli’ hissediyor
PKU Hasta Araştırması: Fenilketonüri hastaları ‘yorgun, üzüntülü ve endişeli’ hissediyor Diyabet hastaları için bilimsel beslenme önerileri ve uyarılar
Diyabet hastaları için bilimsel beslenme önerileri ve uyarılar Fenilketonüri hastalığı doğru beslenme ile önlenebilir mi?
Fenilketonüri hastalığı doğru beslenme ile önlenebilir mi? Protein nedir? Hangi besinlerde bulunur? Faydaları ve zararları
Protein nedir? Hangi besinlerde bulunur? Faydaları ve zararları
 Protein nedir? Hangi besinlerde bulunur? Faydaları ve zararları
Protein nedir? Hangi besinlerde bulunur? Faydaları ve zararları
YORUMUNUZ VAR MI?
Bende bir PKU’lu olarak bu yazıyı çok sevdim ve bilgilendirici buldum arkadaşlarıma kaynak olarak vereceğim teşekkürler.